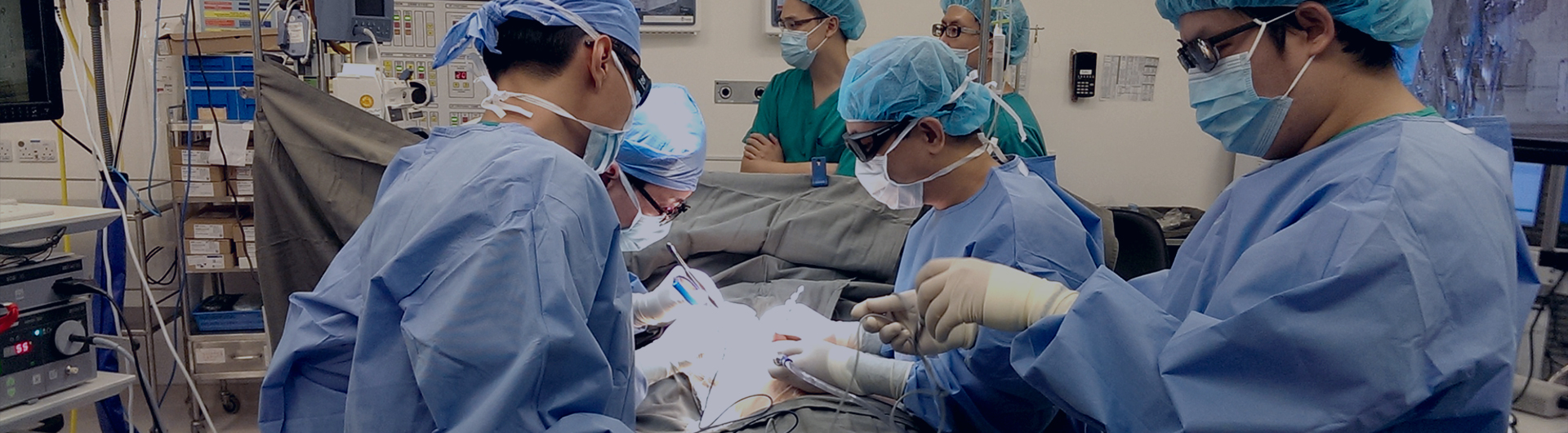
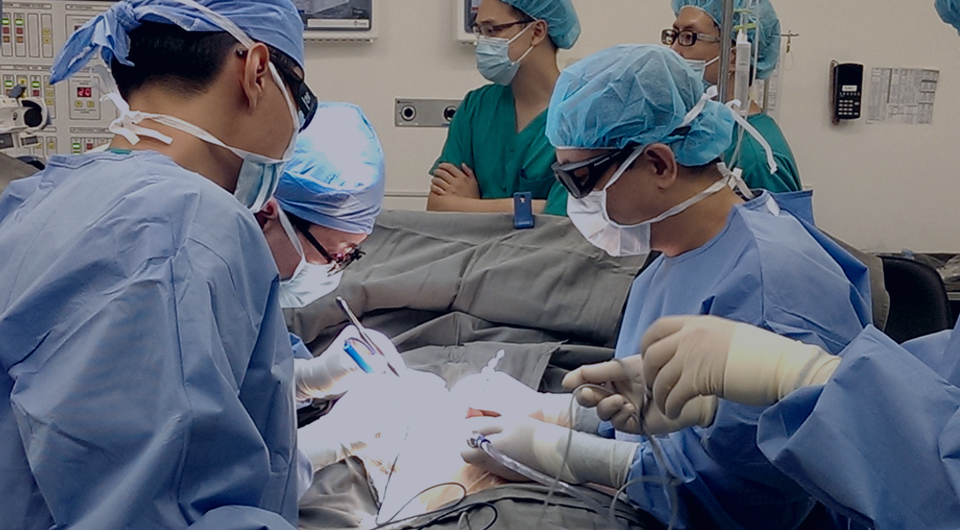
疫苗故事 | 路易斯·巴斯德--狂犬疫苗的发明人
在19世纪,狂犬病每年都要夺走数以百计法国人的生命,由于当时没有疫苗,也没有免疫球蛋白,对付狂犬病,人们只能使用烧红的
铁棍烙烫伤口。因为欧洲人相信,火焰与高温可以净化一切事物,包括肉眼所看不见的细菌。
当时只要是被动物咬伤的人们,都会被村庄中的壮汉们强压至打铁铺,请铁匠用烧红的铁棍去烙烫伤口,想藉此“烧”死看不见的病原
,但如此原始、残酷的作法,并没有办法治疗狂犬病,常常只是加速死亡的来临。
一个名字叫巴斯德的人改变了这一切!
一百年前,在法国,一位大学生登上了一列火车,邻座是个看上去像农民的老人,老人手执念珠(背诵圣经的工具)嘴里念念有词。
“先生,你还相信这些过时的东西?”学生问。 “是的,我相信。你不信吗?”老人回答。 学生笑了笑:“我不相信这些愚昧的事情。听
听我的建议,把你的念珠扔掉,了解了解科学对此的解释。”
“科学?我不懂这个科学,也许你能给我解释解释。”老人说。 学生说:“这可不是一两句话就能说清楚的,请留下你的地址,我会
寄给你一些书,你自己看吧。” 老人从内衣口袋里拿出一张名片,学生接过一看,脸一下子就红了,低头不语。 名片上写着:路易士·巴
斯德,巴黎科学研究院院长。
路易士巴斯德(Louis Pasteur)1822年12月27日出生于法国东尔城,毕业于巴黎大学,信仰基督教,法国著名的微生物学家、化学家
,1895年9月28日逝世。近代微生物学的奠基人。像牛顿开辟出经典力学一样,巴斯德开辟了微生物领域,他也是一位科学巨人。
巴斯德曾任里尔大学、巴黎师范大学教授和巴斯德研究所所长。在他的一生中,曾对同分异构现象、发酵、细菌培养和疫苗等研究取得
重大成就,从而奠定了工业微生物学和医学微生物学的基础,并开创了微生物生理学,被后人誉为“微生物学之父”。美国学者麦克·哈特
所著的《影响人类历史进程的100名人排行榜》中,巴斯德名列第12位,可见其在人类历史上巨大的影响力。其发明的巴氏消毒法直至现
在仍被应用
1882年,巴斯德被选为法兰西学院院士,同年开始研究狂犬疫苗。狂犬病是一种可怕的传染病。人和家畜被病犬咬伤之后,也会患狂
犬病。它每年要夺走数以百计法国人的生命,当时没有疫苗,也没有免疫球蛋白,对付狂犬病,人们只能使用烧红的铁棍,19世纪的欧洲
人相信,火焰与高温可以净化任一切事物,包括肉眼所看不见的细菌。
当时只要是被动物咬伤的人们,都会被村庄中的壮汉们强压至打铁铺,请铁匠用烧红的铁棍去烙烫伤口,想藉此“烧”死看不见的病原
,但如此原始、残酷的作法,并没有办法治疗狂犬病,常常只是加速死亡的来临。
1880年底,一位兽医带着两只病犬来拜访巴斯德,请求帮助。能不能制成狂犬疫苗呢?1881年,巴斯德组成一个三人小组开始研制狂
犬病疫苗。他们希望从采集病犬的唾液进行研究,但这种采集十分危险。巴斯德和他的团队冒着被咬伤的危险采集狂犬的唾液,一次,巴
斯德为了收集一条疯狗的唾液,竟然跪在狂犬的脚下耐心等待。
他们将采集的狂犬唾液注射到健康犬只的脑中,健康的犬只果然马上发病死亡,历经过数次的动物实验,巴斯德推论出狂犬病病毒应该
都集中于神经系统,因此他大胆地从病死的兔子身上取出一小段脊髓,悬挂在一支无菌烧瓶中,使其“干燥”。他发现,没有经过干燥的
脊髓,是极为致命的,如果将脊髓研磨后将其和蒸馏水混合,注入健康的犬只体内,狗必死无疑;相反的,将干燥后脊髓和蒸馏水混合注
入狗的身上,却都神奇的活了下来。
巴斯德于是推断干燥后脊髓的病毒已经死了,至少已经非常微弱。因此他把干燥的脊髓组织磨碎加水制成疫苗,注射到犬只脑中,再让
打过疫苗的狗,接触致命的病毒。经过反复实验后,接种疫苗的狗,即使脑中被注入狂犬病毒,也都不会发病了!巴斯德高兴的宣布狂犬疫
苗研发成功!
1885年,一位几乎绝望的母亲,带着被狂犬咬伤的9岁小男孩约瑟芬(Joseph Meister),来到了巴斯德实验室门口,哀求巴斯德救救
她的孩子。为了不眼睁睁看着男童死去,巴斯德决定为约瑟芬打下人类的第一针,这时距离约瑟芬被狗咬伤已经四、五天了;巴斯德在1 0
天中连续给少年注射了十几针不同毒性的疫苗。
在治疗约瑟芬的过程中,有人提出:“把孩子当试验品是不道德的,我们不知道应该用在人身上的剂量。”巴斯德的回答则是:“我确
定我是在救一个孩子的命,而不是在试验我的疫苗。”
他为小约瑟芬彻夜难眠,怕自己害了他,甚至在回家乡阿尔布瓦休息前,还叮嘱小约瑟芬每天写信告诉他发生的事。15天假期里,巴斯
德家里连续不断地收到病人汇报情况的信件。一个月后,少年健朗如常,安然返回家乡。巴斯德成为世界上第一个能从狂犬病中挽救生命的
人。
消息传开,国内外络绎不绝的患者蜂拥而至前来注射疫苗,狂犬疫苗取得了空前的成功。巴斯德和助手日夜忙碌。长年的过度工作,严
重损害巴斯德的健康。1887年10月23日上午,他脑溢血又发作了,倒在写字台上,舌头麻痹,说不出话来。
在狂犬病疫苗研究上的努力使得巴斯德更加赢得法国民众的尊重,1888年,法国政府为表彰他的杰出贡献,成立了巴斯德研究所,他
亲自担任所长。1889年,生产工艺已经比较成熟的狂犬病疫苗正是由巴斯德研究所推向市场,并开始广泛使用。
1895年9月28日,也就是他72岁时,他在亲友及学生的环绕中在维伦纽夫.勒伊丹(Villeneuve L’Etang)去世。差不多有半个多世纪
,他都是在半身不遂的情况下,在科研领域拼命研究往前冲的。
巴斯德研究鸡霍乱、炭疽杆菌以及狂犬病上的指导思想日后成为免疫学的基石,其他科学家应用巴斯德的基本思想先后发展出抵御许多
种严重疾病的疫苗,能够预防斑疹伤寒和脊髓灰质炎等疾病。
对于自己所取得的成就,巴斯德归功于神。巴斯德是名敬虔而保守的基督徒,他认为科学与基督信仰并无矛盾,并坚信科学拉近人与神
的距离。他说:“对大自然越有研究,就越感受到造物主奇妙的工作”,“科学驱使人更亲近神”,“如果承认上帝的存在,这一个信心实
比一切宗教的神迹更为超奇,不可思议。如果我们有了这种信心,这种悟性,那便不能不对上帝下跪敬拜了。”
巴斯德时常在在实验室里一边工作,一边祷告。他相信,上帝由于其无限的恩慈,不可能为人类创造一种惩罚的灾祸后,不同时为人类
造出一种解药来。巴斯德说“我对大自然愈有研究,就愈感受到创造主奇妙的工作。我知道得越多,我对耶稣的信仰就越接近一个乡下农夫。”
当时在欧洲知识份子中,流行的是“自然发生论”,认为生命可以由没有生命的物质中自然产生,而达尔文进化论中的“物种起源”,
更被“自然发生论”拥为经典。对此,巴斯德予以坚决反对。他相信生命是神所创造而非自然产生的,认为如果物质本身可以产生生命,那
生命 (的价值)变成短暂,物质反成永恒。他坚信生命(而非物质)才有永恒的价值,他并以准确的实验(天鹅颈烧瓶实验)证实了这一点。
巴斯德年迈时曾回自己的母校演讲,并谈到了他能够在科学领域节节得胜的两大要素:一个是信心,相信神的启示。二是热忱(enthusiasm)。
“这个生词是由En及Theo合成,En是里面,Theo是神。真正持久的热忱,是来自神在我心里。”或许,巴斯德所言正是一个真正希冀在科
学上有所建树以及使用其科学成果之人或企业所应该秉承的。
本文转载自 新苗 生物制品圈
侵权联系删除
敌人之友:类固醇和美国(美国类固醇简史)
在体育运动中,除了职业运动员使用类固醇或增强性能的药物外,可能没有其他话题能引起人们情绪上的反应。对于某些人来说,使用
药物是正当的,但是对于某些人来说,使用任何药物都不利于公平竞争。考虑到底是否正当这一点,所以今天的文章我将着眼于美国的类
固醇史,特别是类固醇的首次使用以及何时成为禁用物质来带大家了解类固醇背后的故事。
类固醇的"发现"可以追溯到1931年的德国,当时化学家阿道夫·布特南德(Adolf Butenandt)找到了一种分离和纯化荷尔蒙雄激素的
方法。匈牙利化学家利奥波德·鲁日奇卡(Leopold Ruzicka)很快改善了这一突破性发现,他发现了一种合成供人类使用的激素的方法。
到1935年,鲁日奇卡和布特南德创造了一批合成的睾丸激素,这是一项科学的进步,因此两人都获得了1939年诺贝尔化学奖。是的,类固
醇的发明者获得了诺贝尔奖。
到了20世纪30年代后期,人类已经将注射睾丸激素用于各种用途,尽管战争来临,但类固醇的发明给人们带来了一种观念,即人类可
以通过化学手段改变战争的想法。在美国,1941年美国队长的诞生非正式地体现了这一点,这要归功于约瑟夫·雷恩斯坦博士(Josef Reinstein)
的秘密血清,使之从瘦弱变得强壮。从而使美国队长成为美国第一个已知的使用了化学增强剂的漫画人物。
除了漫画英雄,到了20世纪40年代,苏联对大量的运动员进行了合成代谢类固醇的使用,这也解释了它们在战后体育赛事中的主导地位。
但是美国很快就了解了苏联可以取得冠军的秘密。到了20世纪50年代中期,美国奥林匹克队医师约翰·齐格勒(John Ziegler)博士找到了一
种方法,很快开发出二氢甲基异黄酮,这就是在健美界更广为人知的美他酮,也被称之为大力补。Ciba制药公司是第一个销售该药物的公
司,到1958年大力补已被FDA批准用于人类。
有趣的是,大力补的第一个已知使用者是美国举重之父鲍勃·霍夫曼(Bob Hoffman)和三个著名的训练者:约翰·格里梅克(John Grimek),
吉姆·帕克(Jim Park)和亚兹·库扎哈拉(Yaz Kuzahara)。这反映了齐格勒当时与霍夫曼和约克杠铃俱乐部之间的密切关系。而更为有趣
的是,格里梅克在未使用大力补之前是著名的举重运动员,也是健美运动的第一批明星之一,所以这也使他对大力补的使用预示了健美运动
的发展。
重要的是,这四名男子的肌肉质量和力量都有所提高,这促使齐格勒在1960年为整个美国奥林匹克举重队使用了大力补,尽管使用了新
的增强化学物质,但美国仍然输给了苏联。更严重的是,在1960年奥运会上,丹麦自行车手克努德·艾内马克·詹森(Knud Enemark Jensen)
在比赛中死亡,这也是首次报道由于兴奋剂导致的死亡事件。克努德死后,媒体开始更多地关注运动中的类固醇。同年,《体育画报》发表
了《我们快乐的毒品运动员》一文,这是一篇有关优秀运动员使用苯丙胺,镇定剂和可卡因的文章。但是不管怎么样,国际奥林匹克委员会
花了7年的时间,直到1968年,才将强制性药物测试纳入奥运会。在此期间,类固醇已成为美国许多娱乐活动(如棒球和美式足球)中公认
的提高性能的手段。
1968年引入的IOC测试效果特别差,但鉴于是历史首次,这段奇葩的历史仍然值得一提。那么差的什么程度呢?在1968年法国冬季奥运
会和1968年墨西哥夏季运动会上,几乎没有人对增强性能的药物测试呈阳性。实际上,第一个被取消兴奋剂资格的奥林匹克运动员根本不是
类固醇使用者,而是五项全能运动员之一的瑞典人汉斯·贡纳尔·里尔恩沃尔(Hans-Gunnar Liljenwall),他在墨西哥奥运会的测试中由于体
内酒精含量超标被剥夺了铜牌。值得注意的是,在同一场比赛中,其他14位运动员对当时未被禁用的镇静剂检测全部呈阳性。
如果在过去的半个世纪中,运动员大量使用了类固醇,那么在接下来的几十年中,组织者将试图揭露此类运动员。1972年,国际奥委会
发起了对所有麻醉品的大规模药物测试,导致7名运动员丧失资格。三年后,合成代谢类固醇被添加到国际奥委会的禁用物质清单中,标志
着公众对该药物的看法发生了巨大变化,但这时候也是类固醇的鼎盛时期。
到了1981年7月,美国铁饼运动员本·普拉内特(Ben Plucknett)的合成代谢类固醇测试呈阳性,失去了其世界纪录,并成为国际田径联
合因其类固醇使用而取消资格的首位运动员。尽管普拉内特受到惩罚,但美国运动员并没有停止服用药物。1983年,委内瑞拉的泛美运动
会陷入混乱,当时出人意料的毒品测试导致数十名运动员退出。包括十几名退出比赛并未经说明返回美国的美国运动员。事情过去不久后,
一场真正的丑闻席卷了北美。
1988年,本·约翰逊(Ben Johnson)成为加拿大自1928年以来第一位在奥运会赢得100米冠军的短跑运动员。约翰逊对合成代谢类固
醇进行了积极的测试,但仅仅三天后,他就失去了奖牌。由于停赛两年,约翰逊的禁令对美国也产生了影响。
到20世纪80年代中期,对于专业运动和非专业运动员中的合成代谢类固醇的使日益增加。人们担心,在他们最喜欢的运动员的刺激下,
青少年和成年人会出于娱乐目的而寻求化学式的增强。
而约翰逊的禁令是美国的媒体的救命稻草,在加拿大人被禁赛之后,美国国会大力游说以研究并检查类固醇的使用。从1988年到1990
年,举行了国会听证会,以确定是否应修订《管制物质法》(1970年)以包括合成代谢类固醇。重要的是,许多此类听证会揭示了类固醇
使用的复杂性。几家机构和医学专家反对这一举动,认为使用类固醇并不会导致该法案中所要求的身体或心理依赖性。医学界的这种混乱与
体育组织代表的一致意见形成鲜明对比,体育组织的代表认为,禁止类固醇将彻底解决体育运动中使用兴奋剂的问题。
经过两年的审议,美国国会通过了1990年的合成代谢类固醇控制法,从而在没有有效处方的情况下将拥有合成代谢类固醇定为犯罪。合
成代谢类固醇与巴比妥类药物,氯胺酮,LSD前体和麻醉性止痛药(如维可丁)列在同一法律分类中。该法律于1991年生效后,非法拥有任
何数量的合成代谢类固醇,即使无意出售或散布,也属于犯罪。这适用于运动员和非运动员,最高可被判处一年监禁和/或最低罚款1000美
元,以起到威慑作用。
在20世纪90年代初期,包括世界摔跤联合会(WWE)和它所包装的明星绿巨人·霍根(Hulk Hogan)在内,许多体育组织都对新规定
感受到很大的压力。并且在这一时期,数千名运动员被揭露在过去曾使用过合成代谢类固醇,这使人们心碎不已。
自20世纪90年代以来,美国对类固醇法又作了几处修正,例如2004年的《2004年合成代谢类固醇控制法》,在控制物质清单中增加了
前体,以及2014年更新的《2014年构思者合成代谢类固醇控制法》,扩大了美国药物管制局(DEA)管制的合成代谢类固醇的清单,使其
包括约二十种新物质,并确立了与虚假标记类固醇有关的新罪行。尽管采取了这种行动,但美国使用合成代谢类固醇和增强性能的药物一直
在继续,健美运动员人数的不断增加也证明了这一点。
对美国类固醇的简要历史进行调查,可以得出两个令人不安的发现。
第一,几十年来,运动员和非专业人士使用类固醇不受惩罚,并且没有任何实际的法律后果。并且在50年的大部分时间里,类固醇在大
部分的时间里都没有受到严格的监管。
第二,关于类固醇使用的立法在职业体育方面基本上是无效的。自20世纪90年代初以来,随着运动员和球队寻求更好的成绩,增强性能
的药物在体育领域的应用比以前更加广泛。而本·约翰逊于1988年在100米短跑中所创的世界纪录已经被所谓的干净运动员击败了好几次,这
一事实让人不禁要问,化学增强是否没有发挥其作用。还是自1988年以来,训练方法有了巨大的改进?
最后一点,我怀疑关于类固醇的谈论有很多都被媒体的狂热所带偏。因为激素替代疗法现已成为公认的医学实践,由于使用了类固醇,
该疗法已帮助无数的男女过上了更好的生活。那么在讨论类固醇时我们会谈论这些吗?不。取而代之的是,我们将敢于承认或被发现为运动
目的服用药物的运动员拿来议论和唾骂。但是如果我们想要真正的了解类固醇的使用,就需要对此问题进行真正的讨论,而不是人云亦云和
持有偏见。
本文转载自百度文章,作者老派力量
抗癌白金——铂类药物的发现趣史
铂类药物,称为“抗癌白金”,可不是“脑白金”那种忽悠式的保健产品,而是货真价实的抗癌药物。它的发现,是一段非常有趣的历
史。
顺铂不是第一个用来治疗癌症的药物,在它被发现之前,已有氮芥类药物、叶酸拮抗剂被用来化疗。但当时化疗的疗效是相当 令人失
望的,癌症不是很快就复发,就是很快产生耐药性。而且化疗更多的是针对急性白血病之类的非实体肿瘤。甚至有医学家断言,实体肿瘤
是化疗的禁区。
顺铂的发现,彻底改变了这一切。
最早发现铂的电解产物,名字叫做顺铂,是一个铂和两个氯基、两个氨基的结合。
它是在1844年最先被都灵年轻的化学家米歇尔•派伦(Michele Peyrone)合成出来。早期科学家总喜欢给自己的发现起名,顺铂当时
也被命名为不伦不类的“派伦的氯化物”。但直到1892年,顺铂才由苏黎世科学家阿尔弗雷德•维尔纳(Alfred Werner)搞清楚结构。限
于科学的不发达,当时化学家们并不知道这玩意到底有什么作用。
派伦合成顺铂之后的120年,在地球另一边——美国,密歇根州立大学生物物理教授巴尼特•罗森伯格( Barnett Rosenberg)正在研究电
场对于细菌分裂的影响。实验很简单,就是在大肠杆菌生长的电解质溶液里加上正负电极通电,最后观察细菌的分裂情况。实验一开始,
就让罗森伯格教授大喜过望:细菌都停止分裂了。看来他的假设——电场能抑制细菌分裂是能够成立的。但是接下去几个月的研究,却让
罗森伯格教授越来越失望:电场抑制细菌生长,仅仅是实验的假象。真正抑制细菌生长的原因,是铂电极在通电后产生的电解产物——顺
铂,抑制了细菌的生长。
令罗森伯格教授意想不到的是,他的失望却给癌症治疗领域以巨大的希望,科学家很快就把顺铂应用于癌症治疗领域的研究。科学家想
的很简单:细菌是一种细胞,顺铂能抑制细菌分裂,那能不能也能抑制癌细胞的分裂、生长呢?
罗森伯格教授的发现是偶然的,其意义不亚于威廉•伦琴发现X射线,亚历山大•弗莱明发现盘尼西林。这些都是科学给人类的意外馈赠。
科学家先把顺铂做动物实验,发现对罹患肉瘤的S-180小鼠有效果。S-180小鼠是当时三类标准的癌症动物模型,另外两类分别是长着腺癌
的CA-755,以及长着淋巴瘤的L1210。有趣的是,后来发现的另一类划时代抗癌药紫杉醇,只对CA-755、L1210有效。
1971年4月,顺铂在美国达拉斯的沃德雷分子医学研究所(Wadley Institute of Molecular Medicine)被用来治疗第一个癌症患者,
显示了一些初步疗效。科学家可能是被试验性治疗结果的鼓舞,马不停蹄,在同年6月就开始了顺铂的临床试验。
经过5年的谨慎实验研究,1974年第一份顺铂抗癌临床证据才问世:顺铂对3/7的睾丸癌患者与7/19的卵巢癌患者有效。要知道之前化
疗对这些实体恶性肿瘤的疗效是空白的,这简直就是从零开始的腾飞!结果令科学家兴奋异常。再结合后来发现的紫杉醇,两种药物同时
使用,抗癌有效性提高到73%。直到今天,铂类药物加紫杉醇,是很多癌症的标准化疗方案。
但是,美国FDA并没有立即批准顺铂上市,因为它的副作用——肾毒性,又给科学家们泼了冷水。很多癌症病人在用顺铂之后产生肾功能
衰竭,总不能治好癌症后肾脏报销了去做透析吧。降低剂量可以降低肾毒性,但是抗癌的疗效就也要降低了。除此之外,顺铂还有消化道
副作用,表现就是恶心呕吐。
这个难题难不倒科学家们。1976年纽约的两名专家不约而同发布了对抗顺铂肾毒性的办法:大剂量的液体输入,利尿药的使用。原理很
简单,就是稀释顺铂的血药浓度、加速顺铂的排泄,在保证顺铂的疗效同时降低肾毒性。治疗恶心呕吐也很简单,科学家到现在已经发明
了很多止吐药,预防化疗引起的恶心呕吐。
于是乎,在解决这些副作用问题后,顺铂1978年先在加拿大上市,然后是美国,后来迅速推广到全世界。顺铂是治疗卵巢癌、睾丸癌、
膀胱癌、肺癌、宫颈癌、头颈部恶性肿瘤以及食管癌、儿科肿瘤的主要化疗药物。
美国人在顺铂的研究中大出风头,同样作为癌症领域领头羊的英国也不甘落后。英国癌症研究所的夏娃•魏尔肖(Eve Wiltshaw)进一
步研究了顺铂之后,发现了铂的另一类化合物卡铂。卡铂就是用一个环丁烷二羧酸取代了顺铂的两个氯基。
因为卡铂的环丁烷二羧酸比原来顺铂两个氯基有更好的稳定性,因此对肾脏的损害更小,同时又能对抗更多种类的癌症(注意,顺铂的
抗瘤谱已经很广了),而疗效与顺铂接近。1980年代在英国首先开始卡铂的临床试验,并于1985年批准上市。
当然,卡铂也有副作用,最主要的是骨髓造血功能受到抑制,验血后会发现白细胞、红细胞或者血小板降低,白细胞降低容易诱发感染,
红细胞降低就会出现贫血、缺氧,血小板降低会导致容易出血。所以化疗病人需要定期验血,及时纠正这三类血液成分的减少。
铂类药物,是科学给人类的丰厚馈赠。只有现代医学普照全球,人类才有可能最终战胜癌症。
参考文献:
1.D. Lebwohl and R. Canetta. Clinical Development of Platinum Complexes in Cancer Therapy: an Historical Perspective and an
Update. European Journal of Cancer, 34(10):1522–1534, 1998
2. Lloyd Kelland. The resurgence of platinum-based cancer chemotherapy. NATURE REVIEWS, AUGUST 2007, 7:573
转载自微博:@希波克拉底门徒 作者:希波克拉底门徒
用炸药治心脏病,究竟是谁想出来的?
我们看电视剧的时候,一般心脏病发作,大家都在找“速效救心丸”。而国外的话就会找硝酸甘油。说到硝酸甘油。那么硝酸甘油是个
什么东西呢?
一说到硝酸甘油,很多人想到的可能是诺贝尔,但是它并不是诺贝尔发明的,其本人也承认了。它最初的发明者是意大利的一名化学家
阿斯卡尼奥·索布雷洛,虽然他发明了这一东西,但是将它视作为恶魔般的存在,因为发现硝酸甘油的过程实在是太惨痛了。
在一次化学实验中,他把甘油加入了含有浓硝酸和浓硫酸的溶液中。不曾想,在搅拌的过程中突然就发出了一声爆炸的巨响。刹那间,
实验室就一片狼藉,很多器皿都被炸破。整个人都炸飞出去几米。万幸的是,他的身体只是受了点皮肉伤,否则真是不堪设想,这时候就又
要提到化工安全了,请珍惜你学化工的男朋友。
化学这一门科目或者说这个行业,做实验的时候出现意外,发生爆炸、火灾都是不可避免的。索布雷洛怎么都想不通为什么会爆炸,于
是本着一名科研人员求真的精神,他决定重复进行之前的操作,虽然这次做好了防护,但是仍旧吓个不轻。
反复经历了爆炸后,他终于发现,引起爆炸的是硝酸甘油,一种只需要将甘油加入硝酸和硫酸中就能轻易获得的物质。并且硝酸甘油的
性质极不稳定,非常容易引爆。所以他在论文中极力强调了它的危险性,让大家不要接触,也包括了后来他的学生——诺贝尔。
也不知道为啥,别的小孩小时候都喜欢玩游戏,可诺贝尔却痴迷炸药,于是他对硝酸甘油产生了强烈的兴趣。他不顾劝阻,整天泡在实
验室里研究他怎么样才可以驾驭硝酸甘油。后来,就是我们知道的诺贝尔发明了炸药,发了大财,但是也因为此不知道牺牲了多少人的生命。
有人说诺贝尔是因为觉得自己名声太差了,所以才需要设立诺贝尔奖来改善自己的公众形象。
不过诺贝尔始终认为发明炸药就是为了和平,只是事与愿违了。到了晚年的时候,他不幸患上了心绞痛。
诺贝尔于1896年突然发病逝世。诺贝尔怎么也想不到,自已打了一辈子交道的“炸药”其实是可以救自己的。
一位英国医生威廉•梅瑞尔有一次准备诊断病人的时候,舌头不经意地舔到了装硝酸甘油的软化瓶子(鬼知道它是怎么舔到的,又不是酸
奶)。没过一会儿,他就感觉到了自己面部潮红,血压下降,还产生了晕厥。他拿自己当“小白鼠”试了几次之后,发现稀释后的硝酸甘油
对身体没有多大的影响。因此,他决定对其他人进行一系列的实验。
该实验一共有35名患者同意参与。他们分别是12名男性和23名女性,年龄12至58岁。每人服用硝酸甘油后出现,都类似于梅瑞尔的症
状,并极大缓解了心绞痛。在这之后,梅瑞尔便大胆地尝试使用低浓度的硝酸甘油来治疗他的患者。
然后于1878年正式提出了硝酸甘油能够治疗心绞痛。
看到效果还不错,梅瑞尔将大量临床试验撰写成论文,并于1879年在《 柳叶刀 》杂志上发表。
恰逢当时欧洲国家顺势疗法盛行,很快这种治疗被广泛使用。尽管临床试验证明是有效的,但没有任何人能够说清楚它的作用机制
什么。直到一个多世纪后,三位美国科学家才发现了硝酸甘油主要的作用机制是产生一氧化氮。到了2002年,科学家才发现实现两者转化
的酶是线粒体醛脱氢酶(ALDH2)。
实际上,心绞痛又称为狭心症(Angina pectoris)是心肌缺血引起的胸痛,一般是由冠状动脉阻塞或痉挛所导致。冠状动脉疾病是心血管
的动脉粥样硬化,为心绞痛的主要原因。而心绞痛也是冠心病最常见的一种表现。当心肌缺乏血液供应时,患者会感到胸前有压迫感。临床
上心绞痛可分为稳定性、不稳定性和变异性三种。
无论是何种心绞痛,发作时最重要的治疗方法是要改善心肌缺血、缓解疼痛症状。
打个不恰当的比喻,就是当我们的道路变窄了,得想办法重新疏通。
而用于药物的硝酸甘油正是发挥这样疏通血管的作用。首先,它能释放一氧化氮(NO),激活鸟苷酸环化酶,使平滑肌和其他组织内
的环鸟苷酸(cGMP)增多,导致肌球蛋白轻链去磷酸化,调节平滑肌收缩状态,引起血管扩张。这样使血管平滑肌松弛。有利于血液循
环,对心血管系统产生益处。
其次,硝酸甘油扩张动静脉血管床,主要以扩张静脉为主。动静脉扩张使心肌耗氧量减少,缓解心绞痛。此外,它对心外膜冠状动脉分
支也有扩张作用。
这也是为什么硝酸甘油能治疗冠状动脉狭窄引起的心绞痛瞬时起效。我们也就不难理解很多人一心脏病发就服用硝酸甘油了。
不过我们要明确一点,硝酸甘油顶多是用来救急的。它并不适合用来长期服用救命。当症状缓解后,还要到医院进行后续的检查、治疗。
有研究指出,人类在长期接触后会对硝酸甘油产生一定的依赖性。相传在19世纪,英国一家炸药厂的多个工人就疑似“上瘾”了。他们平时
干活都没事,但周末放假休息时就有心脏病。甚至有几个工人突然在家里猝死了。
相关机构也立即派人前往工厂展开细致的调查。结果发现,他们很可能是习惯了吸入硝酸甘油尘粒。一旦离开工作场合,工人就会产生
不适,才有了“周末心脏病发作”的蹊跷情况。而原本可能有冠心病的人,则是因为突然失去药物来源而身亡。因此,硝酸甘油化身为“救
命药”并非万无一失。对硝酸甘油来说,无论是炸药还是救命药,都是意外发现的。如今拿它来杀人抑或是救人,则取决于人类的一念之间
了。
乙肝疫苗研制用了17年,改变历史的疫苗是如何诞生的
人们很久没有像现在这样热切地期待一款疫苗了。没有特效药,传染性强,似乎只有疫苗才能真正制止新冠病毒在人间流行。进入6月,各种关于疫苗的新进展让人感觉希望就在眼前。世卫组织紧急项目技术主管称,全球已经有超过120款候选疫苗正在研发,而实际数
字可能更多。国资委日前发布消息,国药集团中国生物新冠灭活疫苗预计年底或明年年初可上市。钟南山也向媒体表露过乐观的态度:目
前中国有5款疫苗进入二期临床试验,到年底,一些紧急的情况将能够得到处理。
如果疫苗真的能够在一年时间里投入使用,这个速度毫无疑问是破纪录的。从历史上看,研制耗时最短的,是诞生于1945年的流感病
毒,从病毒被发现到疫苗诞生用了12年;乙肝疫苗用了17年;比较长的是麻疹病毒疫苗,用了46年。直到去年,登革热病毒疫苗才被研
制出来,此时,距离病毒被发现已经过去59年。很多病毒至今令人束手无策,比如,单纯疱疹病毒发现96年来,疫苗始终没能诞生;还
有一个世纪难题——艾滋病病毒,无数科学家前赴后继地努力着,但被发现37年来,依然没有一款疫苗对它有效。
如果把这一串数字视为某种规律,那么新冠疫苗在一年之内被研制出来并投入使用的概率似乎很小。但在默沙东新药研究院工作多年
的一线科学家梁贵柏表达了他的乐观:“我们现在的疫苗研发技术比以往任何一个时期都先进很多,有很多新的方法和技术可以应用。”
每一种药物都有自己的故事,疫苗也是如此。在新出版的《新药的故事2》中,梁贵柏写了从癌症化疗药物到新兴的单克隆抗体等药
物研发的故事,其中也提到流感疫苗。
新书付梓时,新冠病毒还在中国蔓延。对当时的舆论环境,梁贵柏最大的感受是:“在自媒体时代,我们迫切需要大量负责任的、不
误导的科学普及信息。”他感叹,现在网络和微信圈子里不实的、不准确的和误导的信息实在太多了。很多健康医药方面的信息,非专业
人士是很难判断真伪的,况且有不少尚在研究阶段,远没有定论。
第一财经:你说我们现在的疫苗研发技术比以往任何时候都先进很多,因而你对新冠疫苗短时间内被研制出来抱乐观态度。具体是哪
一项技术能缩短疫苗研制时间?
梁贵柏:首先我要声明,疫苗研发不是我的专业,具体技术细节我并不是很了解。我认为,现在研发疫苗的进展主要是在遗传工程方
面,也就是核酸技术。只要知道了病毒的表面抗原蛋白,合成它的基因,就可以用遗传工程的方法把它在体内表达出来。这种技术现在已
经相当成熟。所以从理论上讲,让人体对病毒表面的某一个蛋白产生抗体应该是做得到的。但是这些抗体能不能杀死病毒,并有可持续的
预防作用,都还没有经过实践的检验。这次新冠疫苗的研发就是对这些新技术的实战考验。
第一财经:人类历史上的几种重要疫苗,它们被研发出来的背景以及过程,是否有相似之处?
梁贵柏:我写新药的故事,每一种新药都有属于它自己故事,但是在底层又有相通的逻辑,以及相似的认知过程,疫苗研发也是一样
的。我的两本书里写过三个疫苗的故事——乙肝疫苗、HPV疫苗和流感疫苗。从研发的角度看,它们有大致相同的流程和步骤,需要经过
相同的安评和临床试验,这些都是绕不过去的“坎”,但是它们各自有不同的技术难关,无法依样画葫芦,简单复制,它们带来的健康和
社会效应也不尽相同。
第一财经:如果要取个平均数的话,研发一款疫苗需要的时间是多久?
梁贵柏:这个很难平均,而且平均也没什么意义。回顾历史,从发现病毒到疫苗上市的时间都在10年以上,但这并不等于是研发的时
间。药企研发疫苗必须有足够立项依据,而不是发现一个新病毒立刻上马。比如埃博拉病毒,发现很多年了,但是我以前没有听说哪家公
司研发埃博拉病毒疫苗,当时既没市场也没有紧迫的健康威胁。近年来被重视了,尽管市场还是很小,但是健康威胁足够大,需要战略性
的投资,所以疫苗项目就上马了,并很快取得了成功。前面我说了,我们现在的疫苗研发技术比以往任何一个时期都先进很多,能不能真
的体现出来呢?考验的时候到了,我持谨慎乐观的态度。
第一财经:你提到市场和紧迫的健康威胁是一家公司研制疫苗要考虑的因素。现在,中国已经基本控制了新冠病毒的流行。作为一名
在医药公司工作的研发人员,你认为,现在研制疫苗是否还有必要?
梁贵柏:从流行病学的角度,人类需要一个安全有效的新冠疫苗,这没有疑问。从经济学的角度,如果新冠疫情在全球出现反复,造
成的经济损失肯定会超过研发疫苗的投入。这是全球的视角。站在中国的视角,也许紧迫性不再像先前那么高,但是为了确保疫情不出现
反复,疫苗还是很有必要的。
第一财经:2017~2018年美国流感大流行,但至今没有疫苗问世。新冠疫苗的研发与两年前的美国流感病毒疫苗研发面临怎样不同
的条件?
梁贵柏:据美国疾控中心估计,那个冬季美国大约有8万人死于流感,是一个非常严重的流感季。因为流感病毒的特殊性,流感疫苗
与我们头脑中传统的疫苗,比如小儿麻痹症疫苗,有很大的区别,有效率不那么高。现在季节过了,同样的流感病毒短期内再次出现的机
率不大,所以不会有很多后续的投入去研发更有针对性的疫苗。
目前美国新冠肺炎的死亡统计略超10万,疫情仍在蔓延,也很有可能会在发展中国家持续扩散。为了保证全球经济尽快恢复正常,发
达国家就必须做这件事,各个研发机构也可以拿到大量的资助,自然会有很多人去做。但研发疫苗毕竟不是做产品,而是做科研,是没法
保证成功的。
本文转载自
《新药的故事 2》
梁贵柏 著
译林出版社 2020年5月版
氯丙嗪:精神病治疗的第一道曙光
在你眼里,是不是每年的诺贝尔生理学或医学奖,都代表了医学的伟大进步呢?那你知道有一年的诺奖发错了吗?直到今天,这个奖项
都被看成是诺奖历史上最大的耻辱。这就是1949年的诺贝尔奖。颁给了一位医生,他发明了用切除病人的前脑叶来治疗精神病的方法。
前脑叶就是大脑的额叶,额叶负责我们复杂的认知行为。比如记忆、思考、决策、表达等等。用手术破坏额叶,精神病病人的狂躁和幻
觉自然也就消失了,病人也就不闹了。但是,一个没有情感、没有判断、没有思维、只有呼吸的人,还能算一个真正的人吗?
有人这么描述这个手术:我的女儿做了手术,完全变成了另一个人,她的身体还在,但她的灵魂却永远消失了。电影《飞越疯人院》的
男主角,就是被强迫做了这种手术。从此,一个活跃的勇士变成了一具行尸走肉。
那么这种血腥残酷的手术怎么能获奖呢?评奖的人当然都不傻。这个手术之所以在当时被错误地颁了奖,是因为在它以前,医学对于精
神疾病的研究进展几乎是0。任何一点有可能的希望都让大家振奋,即使是一种今天看起来惨无人道的外科手术。
精神病:最后的黑暗之地
广义的精神病包括精神分裂症、抑郁症、情感障碍等很多种,狭义的精神病专指精神分裂症。这类病人可能出现幻觉、妄想、行为和情
感异常。这节课提到的精神病,指的就是狭义的精神病,也就是精神分裂症。
根据推测,历史上很多名人都有过精神障碍。比如梵高、莫泊桑、果戈理,以及1994年诺贝尔经济学奖得主纳什。在以前,人们对于
精神病充满了无知,对病人蔑视、反感、厌恶,甚至残害。远古时代,人们以为精神病是鬼进入了人体。所以,就给这些病人脑袋上打洞,
以为这样可以让“恶魔出逃”。
到了20世纪30年代,有的医生给精神病的病人注射胰岛素,让他们低血糖、昏迷,用这样的办法来治疗病人的躁狂、幻想。再后来的
治疗办法,就是咱们刚才说的,能让病人变成行尸走肉的外科手术。别看这种手术残忍,这在当时可是有钱人才能做的手术。没钱的人就
只能被关在疯人院,给他们戴上手铐脚镣,或者捆起来。最“人道”的方法,是把病人关在阴暗、狭小、肮脏的地下室里,让他自生自灭。
这就是当时精神病病人真实的生存状况。
人们只看到了病人攻击、逃避、躁狂、抑郁、自残,甚至自杀的外在行为,但是他们的内心却是错乱、颠倒、虚幻和恐怖的黑白世界,
没人能了解。直到一个药的出现,精神病病人的生存状况才发生了改变。这就是氯丙嗪。
氯丙嗪:意外所得的治疗革命
这个药你可能很陌生,听起来好像没有青霉素、疫苗、DNA这些发明如雷贯耳。但是,《英国医学杂志》(The BMJ)却把这个药评
为1840年以来,医学领域的15个重大进展之一。在我看来,氯丙嗪的发明可以和青霉素相媲美。因为,它打开了精神病病人身体和心灵的
双重枷锁,让医学的光芒照亮他们的世界。氯丙嗪是医学发展重要的里程碑之一。
下面我们看看它是怎么诞生的。战争的时候很多伤员会失血,甚至休克。1949年,法国的一位军医亨利·拉伯里特(Henri Laborit)一
直都想找一种抗休克的好药。他找到了一种药,但是这个药抗休克的效果并不好。
受了重伤的人会焦躁不安,拉伯里特给他们用了药以后,发现这些人居然都安静下来了。他想,这个药是不是可以改变人的精神状态呢?
拉伯里特立刻把这个发现发表了出来。放到今天,这样的文章可能直接就被编辑扔了。因为他只写了观察到的现象,全文连靠谱的数据都没
有。但是在当时,还真的有制药公司相信了。这个公司加紧研发,很快就在拉伯里特用的药基础上,改良出一种新药。毒性更小,作用更强,
这就是氯丙嗪。
1952年,医生把这个药用在一个躁狂症的病人身上,这个病人的症状很快就消失了。吃了一段时间后,他的行为甚至接近正常人了。又
过了1年,氯丙嗪在法国上市。1954年,氯丙嗪在美国上市。10年后,全世界有大约5000万的精神病病人用上了氯丙嗪治疗。
大量病人的症状得到了缓解,其中很多病人甚至可以恢复社会功能,自力更生。得了精神病的人不一定非得住院,很多人在家吃药就能
控制症状。这在全世界,掀起了一场精神病病人的“非住院化运动”。就拿美国来说。1955年住院的精神病病人是55.9万人,10年后降低到
了45.2万人。住院人数减少了近20%,有的精神病医院甚至由于缺少病人而关门了。
精神病药物的治疗革命
在氯丙嗪出现前,人们认为疯了就是疯了,就是废人了。有了氯丙嗪,近75%的急性患者可以重新融入社会,参加工作,正常生活。慢
性患者也有相当一部分的病情得到了控制。氯丙嗪开启了药物治疗精神疾病,让病人恢复社会功能的新时代。
1989年氯氮平问世,1993年利培酮问世。这两个药代表着抗精神病药物进入第二代。到了2002年,人类又开发出第三代抗精神病用药
阿立哌唑。在我看来,氯丙嗪带来的进步和意义,已经不仅是个药,不仅是一种治疗精神病的物质载体了。它给医学带来了三个进步:
第一个进步是,氯丙嗪的出现,赶走了所有不科学的解释。
很久以前,人们把精神病的病人看成是魔鬼附体。到了公元前400-500年间,希波克拉底把病人从神鬼的桎梏中解救出来。希波克拉底
认为这些病人身体里没有鬼,是人自身出了问题。再后来,人们把精神病归因于道德,这又给这种病打上了一个“羞耻”的烙印。笛卡尔的
二元论则认为,精神是独立于物质存在的。
这些误解都源自于人们不了解精神病的发病机制。很多病,医学可以通过客观的检查找到明确证据。比如,冠心病可以做心电图,做造
影,做冠脉CT,那么精神病病人的幻觉拿什么来精确检测呢?咱们只知道是大脑出了问题,但是病人外表看不出任何异常,即便去世后尸
体解剖也找不到异常部位。所以,关于精神疾病就充斥着各种不科学的解释。
直到抗精神病药物的出现,病人只要吃上药,很多症状就可以得到缓解。所有疾病一定有物质基础,找不到不代表没有。这一理论再次
得到夯实。所有不科学的解释不攻自破。不需多说,也不需要辩论,事实胜于一切雄辩。
第二个进步是,氯丙嗪的出现,拓展了医学的治疗思路。
有些病,咱们只要了解确切的机制,了解病因和疾病的因果关系,就可以治疗。比如,有些肺炎可能是因为某种细菌感染导致的。那么
找到了细菌,就可以用青霉素治。青霉素干扰细菌细胞壁的合成,让细菌死亡,然后依靠人体免疫细胞自我修复,治愈疾病。
但是到目前为止,更多的病找不到确切的因果关系和发病机制,这些病怎么治呢?比如高血压、糖尿病,比如咱们今天讲到的精神病。
难道必须等到搞清楚因果关系的源头,才能开始治病吗?
1972年,也就是氯丙嗪上市19年的时候,医生发现氯丙嗪的作用和大脑内的多巴胺受体相关。氯丙嗪阻断多巴胺受体,对大脑内的神
经递质进行干预,就可以减轻精神错乱。也就是说,我们可能短期内没办法精确了解因果,但只要能找到发病的某个因果链条,阻断链条,
同样可以治病。从干预因果,到干预因果链条,这是医学治疗思路的拓展。
1957年,人类研发出第一个抗抑郁药。《2014年全球精神疾病药物研发报告》称,目前全球生物制药公司在研的精神疾病药物共有
119种。其中,精神分裂症药物36个、抑郁症药物29个、注意力缺陷障碍和多动症药物15个、抗焦虑药物15个、自闭症药物6个。这些药
的原理,都是作用于发病中的某个因果链条,或者某种递质。
2019年3月,美国FDA审批通过新型抗抑郁药右旋氯胺酮,它的作用原理还是针对某个因果链条,作用于某种递质。也就是说,虽然迄
今为止,很多病的确切发病机制我们还没办法彻底搞清楚。但是为了让病人的病能治,为了让病人好好地活着,那么只要了解了其中的某个
关键环节,同样可以起到很好的疗效。这何尝不是一种伟大的进步呢?
第三个进步是,氯丙嗪的出现,让医学的人道主义照亮最后一片黑暗之地。
在氯丙嗪之前,医学几乎在各个领域都有了长足的进步。但是,精神疾病依然是荒芜一片。想象一下,在这个医学没有取得突破的领域
,那些被关进疯人院,或者走失在人间的精神病患者,他们被看成贱民、恶魔,过着悲惨的生活。
医学的光辉从来没有照亮到这片庞大的、黑暗的区域。很多人对于这类病人充满了蔑视,有些人甚至去恶意攻击他们。也许,我们在街
上偶尔遇到走失的严重精神障碍的病人,他们满身污秽,在街边翻垃圾找东西吃。会不会有人告诉自己的孩子,这是疯子,离他远点呢?
可是,他们是病人,他们没有道德问题。也许有一天,我们周围可能就有至亲的朋友或者亲人会发病。
氯丙嗪,第一次让医学的曙光几乎照亮了疾病黑暗世界的每一个角落。从此,很多精神病的病人可以像正常人一样活着。医学,让人文
更人文。
划重点
1. 精神和肉体不是两个完全独立存在的东西。
2. 从控制因果,到阻断因果链条,这是医学对疾病认知的进步。
3. 氯丙嗪是科学的人道主义。
思考题
精神病是一种疾病,并不是过去大家以为的鬼上身。你身边是否曾遇有过精神病患者?
我有一个同事犯过精神病。那年我们19岁,参加工作不到一年,这个女同事喜欢上了一个兵哥哥,他们是笔友。在用笔恋爱的过程中,
不知道什么原因致使她忽然患病了,好像是双方起了争执,家里人赶紧把她送进了成都的第四人民医院——精神病院。那是80年代末期,
大家对这种病没有一个认知,觉得很可怕。她家里也没有这个病史。在医院呆了1个月,并且把这个男生从部队找来,物理治疗和心理治疗
双管齐下,一个月后她就好了,然后来工厂上班,自己挺不好意思的,但我们大部份同事倒没有看不起她,还和以前一样的对待她,慢慢也
就恢复正常了。最后这个男的转业了,还和他结婚了。他们结婚生了孩子,一切切就正常了。
精神疾病值得我们高度警惕,根据卫生部数据:中国精神疾病的总发病率约为17%,差不多每8个人当中,就有一个精神疾病患者。这
么高发而且可能带来严重后果的疾病,却没有得到应有的重视。 精神疾病并不是我们通常所说的想不开,所以也不是喝顿大酒,出去散散
心就能解决的。精神疾病不但需要服药,就像薄医生介绍的这些,而且严重的要住院,通过各种当时综合治疗。 如果你长期失眠,味觉减
退,对干什么都提不起兴趣,最好赶紧去医院的心身科看看,少千万不要自己扛着。目前医院在这方面的治疗已经十分完善,不少人坚持
治疗症状会有明显改观,并且在一段时间后完全治愈。
本文转载自:简书 作者:满塘荷叶一枝莲
【研发小故事】百浪多息:从染料到药品的进化之路
本文转载自春秋君 药春秋 2022-01-02 22:13
百浪多息 Prontosil
百浪多息(英语:Prontosil)是世界上第一种商品化的合成抗菌药(Synthetic Antibacterial Agent)和磺胺类抗菌药
(Sulfonamide antibacterial),它的发现和开发开启了合成药物化学发展的新时代。
可能百浪多息在非医学领域外不是很著名,很多人听到这名字的反应是:这名字很文艺,但它是药,而且还是种红色染料,不,应该
说它原本是染料,后来被捣鼓成了药,百浪多息作为人类合成的第一种商业化抗菌药,在抗细菌疾病史上意义非凡。
百浪多息的发现过程漫长且曲折,下面春秋君给大家讲一下百浪多息是怎样从染料变成救命神药的。
20世纪的时候,医生们对于细菌感染性疾病无计可施,很多时候都是眼睁睁看着感染导致的并发症带走病人的生命。
但是,人类之所以能一直发展,是总有人能挺身而出,解决难题,他们想法超前,推动历史前进。
保罗·埃尔利希便有这么一个想法:研究出一种只攻击病原体而对人体没有危害的药物。
他先是发明了结核菌的抗酸染色。有一次,在染色过程中,一个惊世骇俗的想法又从他脑中蹦出,有没有可能存在一种物质,“赖
上”(附着)特定的病原体,“吸干”它们的“血”(杀它)。
后来,他研究出了编号“606”(砷凡纳明)的药物,可以有效治疗梅毒,但只能用来治疗梅毒,而那个心中的药,他穷极一生也没找
到。
历史总是由很多人推动的,如果一个人没法推动,也总有后继者,这就是人类历史不断发展的原因。
于是格哈德·多马克就出现了。
多马克在一战的时候是一名医疗兵,志在当医生,但因为见到太多伤口感染去世的人,深感无能为力的他转向药物科研的领域,而科
研资金短缺让他连养家糊口都成问题。
在他山穷水尽的时候,转机来了,传说中的伯乐找到了他,也就是拜耳公司的海因里希·赫连。
海因里希.赫连是苯巴比妥的发现人之一
苯巴比妥是一种巴比妥类的镇静剂及安眠药
拜耳公司当时已经研发出了一些抗寄生虫药,如日耳曼宁和扑疟奎宁这两种抗锥虫和疟疾的药物,但是面对细菌依然束手无策。
赫连同意多马克文章中的观点:直接杀菌的物质会伤害到人体本身,应该寻找一种让细菌变弱的物质(后来的抑菌剂),再让人体免疫系
统清除它们。
经过努力,多马克成功建立了链球菌感染动物模型。
但是他的团队试验了上千种化合物,都失败了。
就在无奈之际,前人埃尔利希"站"了出来,他当年发现红色的偶氮染料对锥虫有效,而多马克想,对抗链球菌或许也有效?
如果说成功是99%的汗水加上1%的幸运,多马克还欠缺那1%的幸运。
就在团队出现分歧的时候,幸运之神说:“差不多了”。
在赫连的提议下,他们将偶氮染料和磺胺基团连在一起,发现这种化合物对感染链球菌的小白鼠有治疗效果,但是还没用于人体试
验。
这时,多马克的女儿意外感染了链球菌,即使是最顶尖的医生也没有办法。多马克别无他选,将还没应用于人体的百浪多息注射进女
儿的体内。
幸运之神微微一笑,多马克赌对了,女儿的病情渐渐好转。
后来,多马克在1935年发表他的研究结果。
至此,百浪多息有了除染料外的另外一个身份—药物,也是第一个抗菌药物。
百浪多息用于人体后,身体会变成红色
多马克的发现拯救了千万人,凭此获得了诺贝尔医学和生物学奖。
尽管后来的研究证明真正起作用的是磺胺,药物的机理清楚后,磺胺药品也逐渐取代百浪多息。但是没有百浪多息,磺胺药品也不知
道什么时候才能被发现,百浪多息拯救了无数人的生命,使得人类在病菌面前终于有了还手之力,这是不可磨灭的。
正如它的名字,历经“百浪”(波折)才被研究出来
出来一阵子便要“休息”(被廉价的磺胺药品取代)
如今,人类已经拥有各种抗菌药和抗生素
抗菌、灭菌已经是轻而易举的事
百浪多息绝对功不可没
▲编辑:Ryan
-The End-
部分内容来源参考:
《抗菌药物前世今生第一回·百浪多息丨华山感染》
Science History Institute: Gerhard Domagk
奎宁:改变世界的药物
奎宁的故事开始于南美洲有记载的历史之前。传说,一个发烧的印第安人,非常的口渴,他发现了一滩死水,并喝了其中的水,他发现
里面的水很苦,并担心自己可能会被水池周围的奎纳-奎纳树给毒死了。然而,他的发烧很快就消失了。他与其他村民分享了这个意外发现。
几个世纪以来,南美印第安人一直使用金鸡纳树皮治疗发烧。
金鸡纳引入欧洲与西班牙殖民者有关。当时,西班牙Chinchon伯爵夫人,她在秘鲁期间感染了疾病,发着高烧,但是被一种树皮的灰烬
治愈。 1638年,她带着树皮返回西班牙,将金鸡纳引入了欧洲,1742 年,植物学家卡尔·林奈 (Carl Linnaeus) 称这棵树为“金鸡纳”
(Cinchona)以纪念她。然而,1941年左右,医学历史学家亚历克·哈吉斯 (Alec Haggis) 拆穿了Chinchon伯爵夫人如何将其带到欧洲的流
行故事。
1570年代,居住在利马(今秘鲁)的耶稣会士药剂师Agostino Salumbrino (1564–1642),发现克丘亚人使用金鸡纳治疗颤抖。虽然它
在治疗疟疾(和疟疾引起的颤抖)方面的效果与其在控制严寒引起的颤抖方面的效果无关,但它是一种成功的抗疟疾药物。此外,Nicolás
Monardes (1571) 和Juan Fragoso (1572) 都记载了利用金鸡纳树皮生产治疗腹泻的饮料。Salumbrino将少量金鸡纳样品送到罗马进行测
试,作为治疗疟疾的方法。在接下来的几年里,金鸡纳树皮,被称为耶稣会的树皮或秘鲁树皮,成为从秘鲁运往欧洲的最有价值的商品之一。
当查理二世国王在17世纪末用奎宁治愈疟疾时,它在伦敦流行起来。
1737年,查尔斯·玛丽·德·拉康达明 (Charles Marie de La Condamine) 发现了治疗疟疾最有效的奎宁形式。1820 年,法国研究人员
Pierre Joseph Pelletier和 Joseph Bienaimé Caventou首先从金鸡纳(可能是金鸡纳)属的树皮中分离出奎宁,并随后命名了该物质。该
名称源自金鸡纳树皮、quina或quina-quina的原始克丘亚语(印加语)词,意思是“树皮的树皮”或“圣树皮”。在 1820 年之前,将树
皮晒干,磨成细粉,然后混合成液体(通常是酒)以供饮用。1850年左右开始大规模使用奎宁作为疟疾预防剂。1853年,Paul Briquet 发
表了关于“奎宁”的文献简史和讨论。
奎宁在欧洲人对非洲的殖民化过程中发挥了重要作用。据说可用于治疗的奎宁是非洲不再被称为“白人坟墓”的主要原因。一位历史学
家说:“正是奎宁的功效为殖民者提供了涌入黄金海岸、尼日利亚和西非其他地区的新机会”。直到第二次世界大战之后,奎宁仍然是首选
的抗疟药。从那时起,其他副作用较少的药物,如氯喹,已在很大程度上取代了它。
奎宁的合成
20世纪初,结构测定还处于起步阶段,人们认为对简单降解产物结构的最终证明需要明确合成具有可疑结构的化合物。在少数情况下,
这可以通过合成天然产物本身来完成,然后与天然产物的真实样品进行比较。因此,具有补充分析的合成通常是一种功利需要的问题,而
不是本世纪下半叶许多伟大的合成化学家的工作所揭示的创造性、优雅的艺术形式。20世纪初,许多研究小组在奎宁的合成或至少重建方
面取得了进展,Paul Rabe的研究小组发表了可能是该领域最重要的成果。1908年,Rabe将辛可尼酮还原为辛可宁,从而实现了一个新的
重要突破,而在1909年,他报道了通过乙醇钠和亚硝酸烷基酯的作用裂解金鸡纳酮,从而产生喹啉-4-羧酸和部醌衍生物。1911年,他通
过用次溴酸处理前者,然后用乙醇钠将所得N-溴衍生物环化脱溴化氢,成功地将辛可毒素转化为辛可尼酮。然而,在第二次世界大战期间
的战时压力下,对其合成生产进行了研究。1918年, Rabe 和 Kindler发表了从喹诺酮中重建奎宁和奎尼丁的合成路线。这是Perkin在50
年前合成奎宁失败后的重大转折点。尽管可用资源不足,但Kaufmann和Rabe的研究小组肯定非常接近完成奎宁的合成。
伍德沃德(R.B. Woodward)从1940年开始担任兰德公司的顾问,1942年,当兰德需要奎宁替代品时,伍德沃德很快解决了他的问题。
这个协会卓有成效,因为兰德还同意资助伍德沃德自己的奎宁合成项目,这是几年前他还是学生时就构思出来的。伍德沃德在他那个时代的
成就是惊人的;它们壮观的性质不仅源于所选合成目标的相关性,还源于他解决合成问题的独创性、他为复杂挑战提供的优雅解决方案,以
及应用这些解决方案所涉及的方法的简单性。
1944年,美国化学家R.B. Woodward和W.E. Doering完成了奎宁合成的前期工作。此后,已经实现了几种更有效的奎宁全合成方法,
但它们都无法在经济方面与从天然来源分离生物碱相竞争。金鸡纳树仍然是奎宁唯一经济实用的来源。
除了化学合成,奎宁生物合成路线也已见报道。奎宁生物合成的第一步中,胡豆苷合酶(Strictosidine synthase)催化色胺和Secologanin
之间发生立体选择性Pictet-Spengler反应,生成胡豆苷。胡豆苷的进一步修饰产生醛。经水解、脱羧从环烯醚萜部分去除一个碳并产生Corynantheal。
然后色胺侧链在氮附近被切割,然后该氮与醛基键合以产生辛可胺(cinchonaminal)。吲哚杂环开环生成新的氨基和羰基官能团。然后氨
基与色胺侧链裂解中产生的醛结合,形成新的喹啉杂环,得到辛可尼酮。最后一步,羟基化和甲基化得到奎宁。
参考文献
1. Achan, Jane, et al. "Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria." Malaria journal 10.1 (2011): 1-12.
2. Kaufman, Teodoro S., and Edmundo A. Rúveda. "The quest for quinine: those who won the battles and those who won the war." Angewandte Chemie International Edition 44, no. 6 (2005): 854-885.
3. https://link.zhihu.com/?target=https%3A//en.wikipedia.org/wiki/Quinine
Cell子刊:砒霜治癌的前世今生:吃最毒的药,治最难的病,「砒霜」抗癌再获新突破
砒霜,是一种闪耀在世界历史与文学作品中的物质,一直被认为是毒药的代名词。在古代,它廉价易得,急性中毒后又没法抢救,因
而被广泛使用,可以说是跨越了阶层、地域与时空的 「经典毒药」。
从《水浒传》中的武大郎,到拿破仑与他的老对手英国国王乔治三世,再到清末的光绪帝。他们的死亡,与砒霜都脱不了关系。
作为一种毒药,砒霜也是各个国家的传统医学中的重要组成部分。在青霉素出现之前,治疗梅毒的一线药物就是砒霜,疯王乔治三世
砒霜中毒的原因,也是因为他患有的精神问题需要砒霜来治疗。
我国文化中自古以来就有「以毒攻毒」的理念,对「砒霜」的药用价值也早有研究。但真正将砒霜入药带入现代医学的,还要从几个
中国学者说起。
砒霜抗癌第一战
1971 年,在哈尔滨医科大学附属第一医院工作的张亭栋从同事韩太云得知到一个治疗淋巴结核的老方,老方中含有用含砷、汞和蟾
毒等多种剧毒物质。这个方子在民间也被用来治疗癌症。
张亭栋也发现这个药方中的砒霜,也就是三氧化二砷(以下简称 ATO),是该方中唯一有效的治疗性分子,汞和蟾毒反而会导致严重
的副作用。
1973 年,张亭栋与韩太云在当地的《黑龙江医药》发表了一篇题为《「癌灵注射液」治疗 6 例白血病初步临床观察》的论文 [1]。
在该论文中,张亭栋与合作者们首次用现代医学的检验方法评估了由 ATO 与汞组成的「癌灵注射液」(癌灵 1 号)对白血病的治疗
作用。这篇一开始没引起太多人注意的论文,却为血癌治疗领域打开了一扇全新的大门。
随后的几年中,张亭栋先后发表了数篇临床研究论文,并将白血病的范围进一步细分到了急性白血病,并报道了使用癌灵 1 号能够实
现完全缓解。
在 1979 年一篇发表在《黑龙江医药》题为《癌灵一号注射液与辩证论治治疗急性粒细胞型白血病》的研究中 [2],张亭栋与合作者
对 55 名急性粒细胞白血病患者的治疗效果及性能进行了分析,发现癌灵 1 号的总缓解率为 70%,甚至有 12 例达到完全缓解。这是
非常惊人的疗效。
随后的十余年中,张亭栋将砷剂的适应症扩展到了急性非淋巴细胞性白血病,并最终发现,砷剂对急性早幼粒白血病(APL)的疗效
极佳,完全缓解率过半。1992 年发表在《中国中西医结合杂志》的题为《癌灵 Ⅰ 号结合中医辨证治疗急性早幼粒细胞白血病 32 例》的
论文 [3],就报道了相关临床实验的结果。该结论后续也得到其他研究的证实。
但遗憾的是,由于所有论文都是发表在中文期刊上,张亭栋的研究并未引起全球医学界的关注。与之形成鲜明对比的,是几乎在同一
时间被国际医学界发现的全反型维甲酸(ATRA)。ATRA 在上海第二医学院王振义团队的研究中,被发现拥有治疗 APL 的优秀疗效,因
为相关论文发表在英文期刊上,也得到国际学术界的广泛好评。
直到 1996 年,ATO 走出国门的事情才出现转机。
走出国门
1994 年前后,刚刚回国不久的陈竺、陈赛娟教授夫妇注意到了张亭栋的研究,并发现其蕴含的巨大潜力。他们认为这样的优秀的药
物值得让全世界知道。两人遂开始合作,对 ATO 治疗 APL 进行了基础机制研究。
1996 年,二人合作在国际血液病顶级期刊 Blood 发表了题为: In vitro studies on cellular and molecular mechanisms of
arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with
downregulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins 的研究 [4],报道了砷剂治疗 APL 的分子机
制。
该研究不仅开启了人类对 ATO 抗癌机制的探索,还终于让这一国产发现走向了世界。
该研究的发表,迅速在国际血液病领域引发了巨大的轰动。Blood 论文发表的一天后,Science 上就刊登一篇题为 Ancient Remedy
Performs New Tricks [5] 的专题报道,将这份中国传统医学的「馈赠」推到 APL 治疗的风口浪尖。
1 年后,陈竺、陈赛娟夫妇与张亭栋携手王振义,在 Blood 上同时发表了两篇题为 Use of Arsenic Trioxide(As2O3)in the
Treatment of Acute Promyelocytic Leukemia (APL): I. As2O3 Exerts Dose-Dependent Dual Effects on APL Cells [6] 与 Use of
Arsenic Trioxide (As2O3 ) in the Treatment of Acute Promyelocytic Leukemia (APL): II. Clinical Efficacy and Pharmacokinetics in
Relapsed Patients 的系列临床研究 [7]。报道了 ATO 在 APL 与 APL 复发患者中的神奇疗效。
这些论文在医学界引起了巨大的轰动,直接将砷剂带向了 APL 疗法研究的最前线。其分量之重,影响之大,从第二篇论文作者列表的
三个中国工程院院士的名字中可见一斑。
砷剂,不只是血癌 随后的二十多年中,无论是国内 ATO 研究的「先行者」,还是世界上其他国家的「跟随者」,都开始对 ATO 的
抗血癌机制展开了研究。也开始注意到砷剂的应用,远不止治疗 APL 这么简单。在张亭栋和他的合作者最早期的研究中,就发现「癌灵
一号」不仅对血癌有奇效,还能够治疗诸如淋巴瘤和食管癌的其他癌症。进入二十一世纪后,各国的学者也发现了一样的现象。ATO 在
部分肝癌和多发性骨髓瘤中表现出一定的疗效。让医学者苦恼的是,ATO 在其他癌症的疗效飘忽不定,并不像它在 APL 中那样能够化腐
朽为神奇。如何让 ATO 更有效的帮助其他癌症的患者,成为了砷剂研究者们下一个需要解决的问题。而精准医疗概念的兴起,为砷剂研
究者们指明了新的方向。
2020 年 12 月 24 号,来自上海交通大学瑞金医学的 卢敏 团队在 Cell 子刊 Cancer Cell 上发表了题为: Arsenic Trioxide Rescues
Structural p53 Mutations through a Cryptic Allosteric Site 的研究论文 [8],报道了 ATO 是如何通过砷原子填补明星抑癌基因 p53
的空腔,以维持后者的稳定,并恢复 p53 抑癌功能的全新机制。这意味着 砷剂在治疗结构突变型 p53 癌症中具有巨大的潜力 。
全世界一半的癌症患者具有 p53 突变,其中带有结构性突变的人群也不在少数。利用精准医疗,现代医学能够快速的识别出适合使用
砷剂治疗的人,并造福广大的癌症患者。该发现也使得砷剂的未来,比 APL 更加广阔。 图片来源:未来科学大奖 2016 年与 2018 年,
砷剂先后获得了欧盟与 FDA 的批准,成为 APL 治疗的一线疗法。在 2020 年,在 ATO 与 APL 研究中作出突出贡献张亭栋与王振义共同
获得了未来科学大奖,表达了中国科学界对砷剂的肯定。我们也由衷的希望,这个来源于中国传统医学的药物,能够拯救更多患者的生
命。
参考文献:[1] 韩太云等,「癌灵注射液」治疗 6 例白血病初步临床观察, 黑龙江医药,卷 3, 1973.[2] 张亭栋 和 荣福祥,癌灵一号注射
液与辨证论治治疗急性粒细胞型白血病, 黑龙江医药,卷 04, 1979.[3] 张亭栋等,癌灵 Ⅰ 号结合中医辨证治疗急性早幼粒细胞白血病
32 例, 中国中西医结合杂志,卷 03, 1992.[4] GQ Chen,. et al. In vitro studies on cellular and molecular mechanisms of arsenic
trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of
Bcl-2 expression and modulation of PML-RAR alpha/PML proteins, Blood, p. 88 (3): 1052–1061., 1996.[5] J. Mervis, Ancient
Remedy Performs New Tricks, Science, 1996.[6] X.-G. S. W. T. S.-M. X. J. Z. X. C. Z.-G. H. J.-H. N. G.-Y. S. P.-M. J. M.-M. L. K.-L. H.
C. N. J. M. P. Z. T.-D. Z. P. P. T. N. K. K. W. M. Guo-Qiang Chen, Use of Arsenic Trioxide (As2O3 ) in the Treatment of Acute
Promyelocytic Leukemia (APL): I. As2O3 Exerts Dose-Dependent Dual Effects on APL Cells, Blood, 1997.[7] G.-Q. C. J.-H. N. X.-
S. L. S.-M. X. Q.-Y. Q. J. Z. W. T. G.-L. S. K.-Q. Y. Y. C. L. Z. Z.-W. F. Y.-T. W. J. M. P. Z. T.-D. Z. S.-J. C. Z. C. Z.-Y. W. Zhi-Xiang
Shen, Use of Arsenic Trioxide (As2O3 ) in the Treatment of Acute Promyelocytic Leukemia (APL): II. Clinical Efficacy and
Pharmacokinetics in Relapsed Patients, Blood,1997.[8] Shuo Chen,. et al. Arsenic Trioxide Rescues Structural p53 Mutations
through a Cryptic Allosteric Site, Cancel Cell, 2020.
本文转载自生物世界
号称迷幻药之王,能做成纸片的毒品-“LSD”是什么来头?
毒品的样子相信很多人都见识过,有粉末、液体、固体、药丸,以及烟草类,但是制作成纸片形状的毒品,相信见过的人不多。然而最
近几年,我国禁毒部门却在多地破获多起贩卖此类毒品的案件。
标题里把这种被称”邮票“的毒品称之为“新型”,其实按时间来算的话,这种毒品可是老爷级,算不得”新型“,早就上世纪60年代
,美国社会就会非常流行。它就是号称迷幻药之王的LSD,一种具有强烈致幻作用的精神类药物。
01
LSD问世于1938年,由瑞士桑多斯公司的化学家艾伯特·霍夫曼 (Albert Hofmann)从变质黑麦中产生的麦角酸提炼合成的一种化学
物质,全名叫做“麦角酸二乙胺(Lysergic acid diethylamide)”。
麦角酸这种物质可以让人产生幻觉,在欧洲中世纪就有记载,有人吃了含有这种物质的变质黑麦面包就会变疯,后人把这种病称之
为“圣安东尼之火病”。后来有人在麦角酸里发现有些成分作为药物使用,而霍夫曼原本也是想从中的找出一种治疗偏头痛的新药,却阴差
阳错的合成了LSD。
经过初步实验,霍夫曼发现LSD没有什么显著的药性,于是便被他扔到一边,一放就是5年。1943年4月的一天,霍夫曼在实验时,不
小心把丢在一旁的LSD粉末误吸入体内,几分钟他就感觉到自己陷入一种异常状态。
眼前出现一连串活动的图像,具有万花筒般的鲜艳色彩;那些在日常生活中一些普通的物品突然变得生动起来,桌椅板凳都似乎有了生
命;听到的每一个声音都像是投在平静水面上的一颗石子,让眼前这些奇妙的色彩产生一圈圈涟漪……
几个小时后,恢复正常的霍夫曼意识这种令人愉悦的幻觉是由LSD造成的,便打算正式试用一次,3天后他口服了大概250微克左右(1微
克相当于百万分之克),原本以为这么少的剂量没多大问题,能产生更令人愉悦的效果,然而这次他却被吓到了。
在与助手骑自行车回家的路上药效发作,霍夫曼感觉天旋地转,周围景物都变了形,自己仿佛是被一面面哈哈镜全面包围。他当时以为
自己一直停留在原地,没有动弹,但旁边的助手事后告诉霍夫曼,当时他骑得飞快。
回到家中,这种怪异的现象愈发厉害,霍夫曼仿佛看到自己的灵魂出窍了,悬浮在空中。而房间里所有物体都变成了怪物,狰狞又恐怖
,邻居和助手在他眼里都不可思议的扭曲着。
他感觉非常焦虑和恐惧,不知道自己能不能从这个怪异的世界回来,甚至担心自己会发疯。幸好第二天醒来他就恢复正常了,也没留下
什么副作用。
但是他的这次自行车致幻之旅却在后来变得十分有名,对多部科幻电影产生了积极影响(猜猜下图像哪部电影里的剧照)。还有好几首
以“自行车”命名的歌曲,比如著名的“女王”乐队就写过一首。
霍夫曼发明的LSD很快就被欧洲生物学界知晓,1947年,瑞士桑多斯公司将LSD命名为麦角酰二乙胺(Delysid),提供给欧洲各研究机构
,后被心理学家们用来研究人脑的病变过程以及用在心理治疗中作为辅助药物。这个时期,LSD的研究还仅限于学术界和少数心理诊所的范
围,普通人并不知情。
02
1949年LSD流入美国,这种这种无色无嗅无味的神秘小分子物质,让当时的中央情报局(CIA)颇感兴趣,开始介入LSD研究。当然他
们并不是用在医学造福人类,而是将其制作成控制思维的化学武器。
这不是CIA的第一次,早在二战期间,其前身“战略情报部”(OSS)在美军上将威廉·多诺万领导下,召集多位神经生理学领域的一流
专家,策划发明一种“审讯辅助药”,让犯人吃后吐露情报。
刚开始,他们试验了多种毒品,最后认定大麻有此功效,通过提纯大麻中的有效成分制作而成,并称为“诚实药”。后经试验发现,这
种药物能让人有强烈的倾诉欲,但很难控制剂量,动不动就让人歇菜(晕厥),实用性非常低。
二战结束后,“战略情报部”(OSS)改名“中央情报局”(CIA),没有停止研制控制思维的化学武器,反而加大力度,特别成立了
一个相对独立的部门,策划一个代号“蓝鸟”的计划(很快这个计划又更名为“洋蓟”)。
CIA把目光盯上可卡因和海洛因,这两种毒品极易上瘾,且停止服用便会有痛不欲生的戒断反应。他们让审讯对象先对这两种毒品上瘾
,然后突然停供,这个时候审讯对象就会浑身疼痛,生不如死,为了求得解脱,便会任人摆布。然而这种方法,很容易就造成审讯对象死亡
,实用性也不强。
了解到LSD药性强烈,微克级的剂量就能让人产生极大的效果,顺理成章的成为CIA又一研究的对象。通过试验CIA发现LSD能让人放松
警惕,他们给一名美军高级官员服用LSD,轻易的就从他嘴里套出一个顶级秘密。药性过后,这位美军高级官员,对自己的泄密行为完全没
有印象,这让CIA大喜,以为自己找到了真正的审讯神药。
然而在进一步试验中,CIA沮丧的发现,LSD很容易引发试验对象产生妄想症,招供的内容更多是“胡说八道”。这也很好理解,LSD
药性很强,很难控制安全剂量,服用者过多服用往往陷入严重的幻觉当中,分不清现实与虚幻,口中的话自然真少假多。
按照惯例,CIA应该要放弃LSD寻找下一个研究对象,但CIA不甘心就此罢手,他们既然LSD能让人暂时变成一个精神分裂症患者,为什
么不用在自己人身上,当作“抗审讯药”用。
比方说深入敌后的特工,一旦被抓,就择机服下LSD,在审讯时,可以“胡说八道”,敌人拿精神分裂人员也没办法。实用性高不高不
是很清楚,但CIA应该没考虑过,特工被抓后如何躲过全身搜索。
冷战时期,CIA不再拘泥于把LSD只做审讯药来使用,他们认为LSD既然能让人发疯,行为失控,为什么不利用这点,给敌对国家的领
导人或是任何持不同政见的公众人物下毒。CIA不仅额计划,更有行动,当时下毒的名单都已经有了,其中就有当时的古巴领导人卡斯特罗
和埃及总统纳赛尔。
随后,CIA发现LSD对每个服用者产生的药效表现大相径庭,不同的场合、不同的心态都会有很大影响。他们希望在非试验室下进行大
规模的人体试验,找出其中的规律。大规模的试验,就需要大量的高纯度LSD,但这难不到CIA。
在后来的CIA解密文档中,有人发现一张高官签署的便条,内容是批准从瑞士桑多斯公司购买10千克的LSD,如此大的剂量,足以让1
亿人发疯一次。
没想到的是,瑞士桑多斯公司直接拒绝了,原因就是他们根本无力生产不出这大数量的LSD。后来才得知是CIA在欧洲的特工搞错了单
位,把微克看成公斤。
没办法,CIA只得在本国找大制药厂研究LSD的合成方法,当时美国大制药之一的“伊莱利利”接受了这个任务,很快他们就向CIA保
证,药厂已经具备生产“吨级”LSD的能力。事实证明他们没有扯蛋,“伊莱利利”在后来成了世界LSD的重要来源之一。
然而直到冷战结束,CIA也没有从LSD身上得到自己满意的结果,却无形中促进了LSD在美国的传播和流行。
03
20世纪60年代美国反文化潮流和嬉皮士运动兴起,有人评价嬉皮士就是婴儿潮时期出生的大学生+LSD。说明在当时LSD随着嬉皮士运
动在美国流行开来。巅峰时期,乔布斯、列侬都是LSD的超级粉丝。
LSD能在嬉皮士流行,不得不提到两个人,一个小说家,一个业余化学家。
小说家叫肯·凯西,你可能不认识,但他写的小说改编成电影,很多人看过。
这本小说的灵感,来自于他在一家对外宣传精神病研究中心,实际是CIA资助的致幻剂研究中心的工作经历。在这里,肯·凯西接触了
LSD,并着了迷。他经常偷吃研究的LSD,以便让自己在“迷幻”的状态下观察研究中心里的精神病人。一段时间后,肯·凯西对精神病有了
一个全新的认识,脑海里有了一个关于精神病院的故事锥形。
他的这本小说叫《飞越疯人院》,出版后,他买了一栋独立的房子,在四周挂了好多音箱,每天放着很响的摇滚乐,除了写作,他最大
的乐趣就是邀请人来这里开LSD派对。在这里,他结识了外号“旧金山名誉市长”的斯坦利,一个业余的化学家,却对LSD却对合成LSD有
很深的造诣。
早就1965年,他与同居女生就已经研究出合成LSD的方法,并制造出了大批高质量的LSD。早期瑞士桑多斯公司制作的LSD颜色发黄,
斯坦利改进了提纯工艺,制作出来的LSD纯净无色。斯坦利是一个LSD教的虔诚者,免费送的LSD比卖出去还多,其目的就是希望更多人享
受到LSD。
在斯坦利的推动下,普通人也很容易买到LSD,不再是大麻独步天下。肯·凯西的派对,斯坦利免费提供LSD,这无疑让很多年轻人趋之
若鹜。不久一群群来自东部的衣着刻板的年轻人来到了旧金山,他们听闻这里有免费的LSD,还有“免费”的女生,便蜂拥而至,当时的美
国媒体称他们为嬉皮士。
到此,LSD与嬉皮士结合,成了相互的重要标志。
04
随着LSD的流行,美国社会开始频繁出现各种因为服用LSD而导致的意外事故、精神崩溃、犯罪和自杀的报告不断增加。因此1966年,
美国加州州长里根宣布LSD为非法药物,禁止合法生产和销售。
LSD在在我国属于一类精神管制药物,由于合成难度高,之前国内并没有LSD多少流通。但近几年,从各地禁毒部门破坏的贩毒案件来
看,这种毒品开始在国内流行起来。
LSD致幻效果特别强,其他毒品的剂量单位都是用毫克,而LSD则需要用微克单位,这也为是其被称为迷幻药之王的原因。
LSD让人上瘾,跟生理上没多大关系,更多的是一种心理依赖。服用者迷恋的是LSD带来的迷幻世界,他们认为,在这种全新的“幻觉”
世界中,闭着双眼也能看到东西,甚至清醒的感觉到自我在慢慢解体,一片一片的“我”正在逐渐融入到整个大自然里去,与整个宇宙达到
天人合一。
但是英国心理医生奥斯蒙德认为服用LSD后的表现完全取决于个人的心理,服用时的环境,周边人物都有可能影响到LSD产生的效果。因
此,并不是所有人产生的幻觉是温和的,更多的是受到心理暗示,产生各种恐怖幻觉。
另外由于长期服用,人的耐受性提高,服用的LSD剂量也不知觉的提升,很容易就造成过量,导致各种意外,人在迷幻状态下跳楼自杀,
甚至残忍的杀人。这也是为什么在流行期间,美国的LSD爱好者发生多起意外事故、精神崩溃、犯罪和自杀事件。
LSD常以口服方式摄入,10微克就可产生明显欣快,50-200微克时便可出现幻觉。由于极易为人体吸收。吸食者服用LSD30到60分钟
后会出现心跳加速、血压升高、瞳孔放大等反应,2到3个小时后产生幻视、幻听、幻觉,对周围的声色、颜色、气味及其他事物的敏感性
畸形增大,对事物的判断能力和对自己的控制力下降或消失。
此时,在生理上常伴有眩晕、头疼及恶心呕吐等症状。当药效消失、迷幻期结束后,吸毒者往往会感到严重的忧郁,由些人还会出现幻
觉重现的现象。对这种现象的恐惧性反应有时会导致自杀行为。
很长一段时间,LSD被称为”疯子药“,因为一些服用者过量使用,精神崩溃或是受到永久损害,成为疯子。
LSD恐怖吗?当然有它恐怖的一面,那为什么要发明,这里就借用发明者霍夫曼的一句话,错用和滥用导致LSD成了我的问题孩子。
最后一个问题:
为什么LSD要制作成指甲盖大小的纸片形状?
第一,为了方便运输和躲避打击,
第二,方便使用者携带,以及服用。
最初毒贩将LSD溶剂滴在纸上,后来就直接把印好的小纸张浸泡在LSD溶液里,这样分布就很均匀,小小的一片纸,含量就有30-50微克
为了能吸引青年人,毒贩们费尽脑筋把纸张设计各种各样的纸制,有抽象艺术和动画图片、纹身花纹,这与邮票风格非常相似,因此吸
毒群体统称为邮票。
目前LSD在国外已经是明日黄花,倒是我国近几年抓获了多起贩卖邮票毒品的案件,贩卖和使用多数是青少年群体,这也意味在LSD在国
内的年轻人群体有泛滥的趋势。
这种毒品毒性极强,一般是摇头丸的3倍。几微克就足以让人产生幻觉,使用后通常会心跳加速,血压升高,并出现急性精神分裂和强烈
的幻觉,造成极大的心理落差。
另外,重庆同济医院戒毒康复中心专家提醒大家要警惕!下面这些都是新型毒品,大家一定要远离,不要去尝试!
近年来,毒品种类不断翻新,冰毒、摇头丸、K粉等毒品已和海洛因一样归入“传统毒品”,涉案毒品已经逐渐衍生出新的种类和形式,
“红冰”“阿拉伯茶”“浴盐”……这些普通人听起来陌生的名字逐渐成为毒品的新名头。
毒品之祸,甚于枪炮
毒品是百恶之首,对人类有着巨大的危害,种类繁多的毒品以其貌似无害的外表吸引着人们,对吸毒本人及其家庭造成巨大的伤害,造
成了极大的安全隐患,毒品是人类的公害,认清毒品危害,防毒、禁毒、拒毒任重而道远,共同向毒品说“NO!”
关爱生命远离毒品
不要让你的梦想因它破碎
一朝吸毒,十年戒毒,终生想毒,毒品会使人格变异,人性泯灭而诱发其他违法犯罪,还会对人体造成巨大的伤害。我们除了为禁毒献
一份绵薄之力外,要做的是远离毒品爱护和调养自己的身心。
比如寻一处“天然氧吧”,漫步于山林间,在这满场的仙气、天籁般的歌声、丹山碧水间的灵秀之气间,看仙境风光,在群山之中、岁
月之巅放牧心灵,感受“神仙地,逍遥游”。
重庆同济医院戒毒康复中心(宣)
历史|人类抗疫史之霍乱
一、何为霍乱
古装电视剧里经常能听到霍乱一词,说到霍乱,相信大部分的人都不会感到陌生。汉朝张仲景《伤寒论》说:"病有霍乱者何?答曰:呕吐
而利,此名霍乱。"但古时候的霍乱并不是现在世界卫生组织所认定的国际检疫传染病。现在我们所说的霍乱(cholera),是由霍乱弧菌引
起、由不洁饮食引起的急性腹泻传染病,患者表现为腹泻呕吐,严重者会脱水休克甚至是死亡。不同于普通腹泻,表现只是拉肚子,霍乱
疫情流行迅速,流行期间发病率死亡率高,危害极大。人类在与霍乱对抗的历史中曾经历七次霍乱大流行,其中第七次霍乱流行中检测出
的埃尔托型霍乱弧菌,具有传播性强的特点,但由于现代科学的进步死亡率相对于之前几次霍乱流行偏低。
人们是如何发现霍乱病菌的传播的呢?19世纪初至20世纪末,霍乱4次出现于英国。1854年,约翰·斯诺(JohnSnow)首次发现霍乱在人
体内繁殖并通过受污染的水传播。[[1]] 1883年,德国细菌学家罗伯特·科赫在埃及成功地分离出霍乱弧菌,并以著名的“科赫法则”确定其
为霍乱的真正病因。直到1884年,霍乱疫苗在西班牙霍乱流行区得到了使用,并在全球推广。
二、霍乱流行从未停歇
据历史记载,霍乱疫情在全球范围内共有七次大流行,1817年在印度尼西亚流行并被发现。自1961年5月以来,不到4年的时间内就传
遍东南亚及西太平洋海域的大多数国家和地区。自1964年后深入亚洲内陆。1970年传入非洲大陆和少数欧洲国家。1973年传入北美洲和大
洋洲。直到1991年霍乱袭扰拉丁美洲,这一年内,就有40万人发病4000人死亡,仅秘鲁经济损失就达到7.7亿美元。
本病在世界范围内已经持续流行59年。世界卫生组织统计2001年非洲霍乱患者占全球的94%。[[2]]据世界卫生组织统计,2014年全球
共出现了14万例霍乱病例,超过2000人因此而死亡。2020年2月15日,霍乱疫情仍在埃塞俄比亚境内肆虐。霍乱仍是全球,特别是发展中
国家面临的一个重大公共卫生问题。霍乱作为世界病,多发于贫困地区,那里卫生环境差,民众饮食卫生无法得到保障,助长了霍乱病菌
的滋生。即使是在发达国家,历史上也受到了霍乱疫情的重击。
影响最大波及范围最广的第七次霍乱大流行至今还未停歇,人类虽然已经与霍乱抗争了 200 多年,但霍乱未被彻底消灭,仍对人类健
康具有威胁。世界卫生组织为此制定了终止霍乱的全球规划图,要求在国家层面实施新的全球霍乱控制战略,到2030年,各国霍乱人数减
少90%,至少20个国家可以在2030年之前消除该疾病的传播,防治霍乱任重而道远。
霍乱大流行时间路径表
霍乱潜伏期短、传播迅速,患者会出现急性腹泻症状,严重者脱水休克甚至是死亡。霍乱杆菌存在于食物和水当中,极易进行传播扩散
,霍乱分为三个时期,泻吐期脱水期以及恢复期,早期症状就是泻吐期的症状,这个阶段患者会出现腹痛腹泻并伴有发烧的明显症状。霍乱
疫情曾在亚洲、欧洲、非洲、美洲都有流行。俄国作曲家柴可夫斯基和其母亲就是在第三次霍乱大流行中感染而去世的。霍乱流行使许多家
庭失去至亲,流离失所。世界卫生组织最新公布的一项研究表明,全球每年约有300万人感染霍乱,其中10万人因此死亡,这两个数字要超
出世卫组织成员每年向该组织汇报的数字10倍。霍乱,是所有人类需要共同对抗的敌人,针对防治霍乱,各国各地区出台了许多措施。
三、全球应对霍乱流行的措施及启示
现代科技与医学的进步,公共卫生条件也较以前改善了许多,霍乱虽然没被彻底消灭,却也不同于以往。疫情流行时造成世界范围内轰
动极大,霍乱在历史上进行的全球流行造成的人员伤亡之惨重,经济损失之巨大是无法估量的。霍乱在各国多次爆发,疫情几经反复。仅对
于霍乱病毒的防治,各国各地区的卫生部门也出台了相应措施来应对霍乱病毒的流行。霍乱的传染性不可小觑,在日常生活中也要注意勤洗
手,注意饮食清洁,吃熟食,不喝未消毒的水。我国在霍乱疾病的研究方面,做了深入的调查研究。目前我国对于霍乱防治的规则日趋完善
,在2015年也仅收到了15个霍乱病例的报告,无死亡病例。世界各地在在政府的统一领导和有关部门密切配合下,有计划有步骤地开展有
关改水、管粪以及垃圾、污物无害化处理和卫生工程建设,从根本上解决好人民大众的饮用水卫生和环境卫生问题。在食品安全的监管上严
格检疫,从源头防治霍乱。由于现有的霍乱疫苗只对部分人有效,并且由于疫苗有毒副作用,一些国家禁止使用。霍乱防治措施主要还是从
改善饮水措施和建立良好的预警系统和完善的报告制度入手。
《霍乱之乱》的作者池莉表示:“我曾经做了三年的流行病防治医生。当我不得不离开卫生防疫专业的时候,我觉得我应该把自己的担
忧写成一部小说:人类尽可以忽视流行病,但是流行病不会忽视人类。我们欺骗自己是需要付出代价的。”人类与病毒的斗争没有停止,遇
到疾病我们不应该去责怪病毒产生的源头,面对病毒,我们都是受害者。究其所以,我们应该对抗的是病毒,而不是人类。
习近平在统筹推进新冠肺炎疫情防控和经济社会发展工作部署会议上的讲话提到:扩大国际和地区合作。公共卫生安全是人类面临的共
同挑战,需要各国携手应对。要继续同世卫组织保持良好沟通,同有关国家分享防疫经验,加强抗病毒药物及疫苗研发国际合作,向其他出
现疫情扩散的国家和地区提供力所能及的援助,体现负责任大国担当。
中华民族历史上经历过很多磨难,但从来没有被压垮过,而是愈挫愈勇,不断在磨难中成长、从磨难中奋起。世界人民在抗击病毒的过
程中也从未停歇,万众一心,众志成城,人类的命运与我们息息相关。面对疫情,我们都应肩负起自己的责任,抛开国与国之间的成见,地
区对地区的歧视。积极响应对抗疫情的举措,从自身做起,从身边小事做起。也只有这样,我们才能更有效地消灭病毒,恢复往日和谐宁静
的生活。
参考文献
[[1]]卢明,陈代杰,殷瑜.1854年的伦敦霍乱与传染病学之父——约翰·斯诺[J/OL].中国抗生素杂志,2020(04):347-373[2020-05-01].https://
doi-org-s.webvpn.wzu.edu.cn/10.13461/j.cnki.cja.006912.
[[2]]罗朝晨, 谢一俊, 张静. 霍乱流行态势及我国霍乱防控中存在的问题%Cholera epidemic situation and the existing problems in
theprevention and control in China[J]. 传染病信息, 2008,021(003):153-154,192.
此作品由科技部引进国外智力管理司推荐
作者:欧宝辛欣
编辑:吴桐
颠茄和阿托品
古希腊神话中斩断命运之线的女神——阿特罗波斯,文艺复兴时期意大利“美丽的女郎”,能置人于死地的草本植株颠茄,有什么联系
呢?正是由于著名的植物分类学家林奈,以阿特罗波斯之名,将颠茄命名为Atropa belladonna,才将她们联到了一起。
古希腊神话中的“命运之线”,在古希腊神话中,有著名的命运三女神——她们是宙斯和法律女神忒弥斯的女儿。这三位掌管万物命运
的女神中,最大的克罗索(Clotho)负责掌管“未来”,纺织生命之线;二姐拉克西斯(Lachésis)决定生命之线的长短,“现在”归她管
理;而最小的阿特罗波斯(Atropos),权力却最大——她掌控死亡,负责剪断生命之线!赋予死亡的她,代表的是“过去”,即使是天父
宙斯,也不能违抗她们的安排。这就是命运三女神:残酷,威严,坚定。
认识一下颠茄,多年生草本,原产欧洲,20世纪30年代引入中国。全草可入药,用于制止盗汗、流涎、支气管分泌过多、胃酸过多等
症状。喜温暖湿润气候,怕寒冷,忌高温。在中国山东、浙江等地有栽培。有茎有叶,有花有果。等一等,小朋友们要注意:浆果千万不能
吃啊!因为吃了颠茄会使瞳孔放大,对光敏感,视力模糊,头痛,思维混乱以及抽搐。
两个浆果的摄取量就可以使一个小孩丧命,10-20个浆果会杀死一个成年人!历史上,早在古罗马时期,著名暴君尼禄的母亲小阿格里
庇娜,就用颠茄毒死了她的丈夫——古罗马皇帝克劳狄一世;而尼禄不仅用颠茄毒杀政敌,更在皇位斗争中毒杀了自己同父异母的弟弟不
列塔尼库斯;而更令人瞠目结舌的是,这母子二人,均“师承”自一位以使用颠茄下毒而闻名的女人洛库斯塔,而由于洛库斯塔本人的多
次下毒事件,被认为是有史料记载以来的第一位连环杀人凶手。颠茄的毒性足以斩断命运之线!直至中世纪,谋杀者也纷纷用颠茄作为毒
药来毒杀他人!
植物分类学家为颠茄命名,一边是斩断命运之线的女神阿特罗波斯(Atropos),另一边是置人于死地的草本植株颠茄,她们是怎样联
到一起的呢?这要归功于瑞典著名的植物分类学家林奈(瑞典文原名:Carl von Linné,1707—1778)。正是林奈,以阿特罗波斯
(Atropos)为颠茄命名——Atropa belladonna。
美丽的大眼睛,Atropa由阿特罗波斯(Atropos)得名,紧随其后的“belladonna”,又是怎么回事呢?“Belladonna” (“贝拉多
娜”)是个俗称,意大利语中,“Bella”是“美丽”的意思,而“donna”则是女郎,合起来就是“美丽的女郎”。林奈为什么会为颠茄
起这样一个名字呢?原来,在文艺复兴时期,意大利曾经有人用颠茄的果实制作女性散瞳的眼药水,取这个名字就是为了纪念这段历史。
用颠茄的根煮水,等水凉后,点到眼睛里,也可以起到散瞳的作用。巧笑倩兮,美目盼兮,眼睛又大又闪亮,的确会让女性看起来更加楚
楚动人,但美丽的背后却需要付出巨大的代价,颠茄制剂会加快心率并且损伤视力,出现远视现象。毒性极强的颠茄终于拥有了自己的名字。
问题出来了——颠茄的毒性为什么如此之强呢?
颠茄的毒性如此之大,1831年,德国药剂师曼恩(Mein)从颠茄的根中分离得到一种生物碱,依然以命运女神阿特罗波斯
(Atropos)为名,这就是——阿托品(Atropine)。
阿托品的命名,古希腊神话中斩断命运之线的女神阿特罗波斯(Atropos),文艺复兴时期意大利“美丽的女郎”(belladonna),能置
人于死地的草本植株颠茄——看似互不相干,最终却由著名的植物分类学家林奈,以阿特罗波斯之名,将颠茄命名为Atropa belladonna,
将她们联到了一起。正是由于颠茄的毒性如此之大,1831年,德国药剂师曼恩(Mein)开始对颠茄进行精确的研究,并从颠茄的根中分离
得到一种生物碱,这种生物碱依然以命运女神阿特罗波斯(Atropos)为名,这就是——阿托品(Atropine)。
阿托品为什么会有如此大的威力?颠茄的根中提取的阿托品,为什么能够如同命运女神一般,斩断命运之线?这,还得从它的作用机制
讲起。
(1)M受体
按照药理教课书上的标准说法,阿托品是M受体阻断药。先说一说“M受体”,“M”就是“muscarinic”的大写英文字头;Muscarinic:
在英文中是“毒蕈碱”的意思;Muscarinic receptor:就是“毒蕈碱受体”,简称为“M受体”。所以,“M受体”就是“毒蕈碱受体”的
意思。
(2)M样作用
通俗点儿说,在我们的心肌、骨骼肌、平滑肌和腺体细胞上有一种蛋白,(还记得《 效应之器——肌肉与腺体的交响曲》里,那三“块”
肉和一根“线”吗?)这种蛋白专门可以和毒蘑菇碱(毒蕈碱)结合。毒蕈碱就如同一把钥匙,插在了位于肌肉和腺体细胞的“锁头”(毒
蕈碱受体,也就是M受体)上。这一插上不要紧,相应的信息就传递到细胞里了:心肌细胞得到信息,开始跳慢了,收缩能力也减弱了(心
脏抑制);支气管平滑肌细胞得到信息,开始收缩了,结果呼吸困难,喘不上来气来了;而胃肠道平滑肌得到信息后,开始收缩,这一下人
可遭罪了,胃肠道平滑肌往上一拱——吐了,往下一挤——拉了,膀胱逼尿肌再一收缩——尿了,结果是上吐下泻,大小便失禁;瞳孔括约
肌收缩的话,会出现瞳孔缩小,甚至是针尖样瞳孔;关键是腺体细胞也来凑热闹,分泌大大增加,人会出现大量出汗(汗腺)、流涎,流口
水(唾液腺)、流泪(泪腺),鼻涕一把泪一把;呼吸道分泌物增加,肺部听诊啰音增强;消化道的腺体分泌增加,消化液增多。
以上所有反应总结起来就是——大汗淋漓、口吐白沫、上吐下泻、大小便失禁,瞳孔缩小、心动过缓等等,这些反应被统称为M样作用
(毒蕈碱样作用)。
(3)M受体阻断作用
阿托品厉害在哪儿呢?别忘了,它可是叫“M受体阻断药”啊!正好能把这些反应全阻断了!全反过来!它就像一把配错的“假钥匙”
一样,也能插进锁头,但是不好使!不好使也就罢了,还占着个地方不让真正的钥匙发挥作用,这样一来,我们刚才讲的“M样作用”就统
统——反过来了!大汗淋漓反过来——汗腺被抑制了,没汗了,不但没汗,身上还发红发热发干了;口吐白沫反过来——唾液腺被抑制了
,不但不吐白沫了,反过来嘴里发干,吞咽困难,还渴!渴!!渴!!!上吐下泻反过来——不吐了,不拉了,大小便也不失禁了,稳是
稳了,太稳了,胃肠道平滑肌蠕动减慢,反过来又便秘了;心动过缓反过来——由于解除了迷走神经对于心脏的抑制,心率开始加快了;
瞳孔缩小反过来——瞳孔开始扩大了;肺部罗音增加反过来——呼吸道分泌物减少了,痰也少了,肺部湿罗音减轻甚至消失了。所有这些
反应加起来可以总结成“阿托品化”反应:
一大(瞳孔扩大);二干(口干、皮肤干);三红(面部潮红);四快 (心率加快);五消失(肺部啰音消失)。
(4)阿托品中毒
凡事都有个限度,对不对?过犹不及,阿托品的用量增加到一定程度,各种反应就会大大增强,出现严重的口干、吞咽困难、声音嘶哑
、面部潮红、皮肤发干、发热、头痛、心动过速、心悸、瞳孔扩大、视力模糊、排尿困难等等表现;进一步影响到中枢神经系统,出现谵
妄、狂躁、眩晕、幻觉、共济失调等等。再发展严重的话,就会出现昏迷、血压下降,最终出现呼吸衰竭而死亡!生命之线被彻底斩断!
所以阿托品的成人常用量:一次仅为0.3—0.5mg;极量为一次2mg!如果用量超过5-16mg,则中毒症状明显;最小致死量为80-130mg,个
别病人为50mg,但在抢救有机磷中毒、酒石酸锑钾中毒、感染性休克时等特殊情况下,用量会比较大!这样一来,我们就知道阿托品为什
么会要命——M受体被阻断了!阻断了!阻!断!!了!!!
(5)阿托品的临床应用
在临床工作中,我们当然不希望药物将人置于死地,我们更希望它会治病救人,发挥应有功效!阿托品有哪些功用呢?——松弛内脏平
滑肌,缓解内脏绞痛。如胃肠绞痛,胆绞痛,肾绞痛等,阿托品可明显松弛内脏平滑肌,有效缓解疼痛。颠茄片的用处还真不小啊!——抑
制腺体分泌,用于全麻前给药,以防止因分泌物过多而阻塞呼吸道。要知道,患者应用全麻药物,在麻醉作用没有消失之前,还不能自主咳
痰。想想看,一旦患者呼吸道内痰液太多又咳不出来的话,就有可能引发呛咳,甚至出现痰液阻塞呼吸道,引发窒息,危及生命,多么严重
的后果!可如果在手术前应用阿托品的话,就可以有效抑制呼吸道痰液的产生,从而避免术后发生痰液阻塞呼吸道的不幸了。——扩瞳、升
高眼内压,调节麻痹,用于验光、检查眼底、治疗虹膜睫状体炎。想想从前爱美的意大利女郎所体验到的吧!还有,M受体激动药毛果芸香
碱应用过量时可以用阿托品对抗。——加快心率,用于治疗缓慢型心律失常。
这主要是由于阿托品解除迷走神经对于心脏的抑制作用,从而加快心率。——改善微循环,用于治疗感染性休克。阿托品可以缓解小血
管痉挛,使回心血量增加,血压回升而改善微循环,起到抗休克作用。对暴发型流行性脑膜炎、中毒性痢疾、中毒性肺炎等所致的感染性休
克,可大剂量使用阿托品治疗。但对于伴有高热或心率过快,则不宜应用阿托品。——解救有机磷酸酯类药物中毒。主要缓解M样作用。
西人眼里颠茄及阿托品皆能斩断命运之线。
不该被遗忘的疫苗先驱,首支腺鼠疫疫苗的发明者瓦尔德玛·哈夫金
纵观人类历史,鼠疫这种烈性传染病曾出现三次大流行,每一次都对当时的世界造成了深远的影响。从公元541年的查士丁尼瘟疫到
14世纪席卷欧洲的黑死病,鼠疫一直是欧洲人挥之不去的梦魔。到了19世纪,鼠疫开始侵袭亚洲,高峰期是在1894年的香港地区和印度,
以及1910一1911年的中国东北。
据记载,东北这场鼠疫最早出现 在俄国境内,于1910年秋天经满洲里蔓延到哈尔滨。疫情暴发初期,哈尔滨每天平均死亡50余人,最
多一天死亡183人。进入冬天后,这场因捕猎早獭引发的灾难更是在中国穷苦百姓聚集地傅家甸彻底失控。不过,这场吞噬6万多人生命的
瘟疫最终被一位名叫伍连德的中国医生成功扑灭,用时不到4个月,这在世界防疫史上堪称奇迹,他发明的防疫模式及工具也沿用至今。
被世界遗忘的疫苗先驱
如第三次鼠疫大流行于1894年从香港开始,通过海路向全世界传播。在瑞士科学家亚历山大·耶尔森(Alexandre Yersin)发现鼠疫杆
菌的基础上,很多学者开始研发人用疫苗,率先攻克这一难题的是乌克兰裔科学家瓦尔德玛·哈夫金(Waldemar Haffkine)。但与之前的
爱德华·詹纳和之后的乔纳斯·索尔克(Jonas Salk)不同,这位疫苗先驱的名字似乎从未真正进入公众的视野。
哈夫金出生于一个犹太教师家庭。1889年,俄国境内大规模的反犹主义浪潮迫使他离开祖国,跟随导师梅契尼科夫(Metchnikoff)来
到巴黎的巴斯德研究所工作。他最初被巴斯德安排参与霍乱疫苗的研究。他研制的减毒霍乱疫苗在印度进行了较大规模的接种,尽管会产生
严重的不良反应,但在一定程度上可预防霍乱的发生。
1896年,另一种烈性传染病一鼠疫深入印度内地,席卷了孟买的贫民窟,其死亡率几乎是霍乱的两倍。当地政府向哈夫金求助,希望
他能研制出鼠疫疫苗。据记载,哈夫金被安排在孟买格兰特医学院的一个临时实验室工作,实验室里有一个房间和一条走廊,工作人员包
括当地的一名职员和三名未经培训的助手。“他没有太多的空间、人力或设施,但这是他第一次独立工作,并拥有自己的实验室。”印度
流行病学家钱德拉坎特·拉哈里亚(Chandrakant Lahariya)说,“他知道,以创纪录的速度开发鼠疫疫苗将使他成为那个时代的领先科学
家。”
1894年3月,哈夫金为加尔各答贫民窟的人们接种霍乱疫苗。他在人们早上上班前开始接种疫苗,并在他们晚上回来后,坐在油灯旁继
续接种。
也许是因为时间紧迫来不及反复传代减毒,抑或是忌惮于鼠疫的强大致死性,哈夫金并没有沿用巴斯德最为擅长以及自己早先开发霍乱
疫苗的减毒思路,而是将分离培养得到的鼠疫杆菌纯培养物在70℃加热灭活了1小时,这就是最初的灭活疫苗。在用疫苗成功地接种兔子后
,他又在自己身上进行了测试,确认除了发热和注射部位疼痛之外,并无其他不良反应。
1897年,哈夫金在当地监狱的“志愿者”中对自己的鼠疫疫苗进行了一次小型对照试验,疫苗的有效性初步得到了证实。在随后四年,
他为成千上万的人接种了疫苗,仅孟买就有20万人,结果非常理想。据统计,灭活疫苗不仅可以预防感染,还可以将受鼠疫影响的社区的死
亡率降低近50%,甚至可以降低接种疫苗时已处于潜伏感染期人群的死亡率。
德雷福斯事件再现
疫苗需求量的不断增长带来了生产规模的扩大。1901年,哈夫金被任命为孟买鼠疫研究实验室的总负责人,手下共有53名员工,负责
制备、分发鼠疫疫苗以及霍乱和伤寒疫苗。作为印度政府的科学顾问,他大力提倡用于预防细菌性疾病的疫苗,成为当地一位颇受欢迎的英
雄。然而就在这时,灾难降临了。
1902年3月,来自旁遮普邦穆尔科瓦尔(Mulkowal)的19名印度村民在接种了同一瓶疫苗后死于破伤风。所有证据似乎都表明,这瓶
编号为53N的疫苗受到了致命的污染。印度政府委员会经过调查,认为疫苗封装瓶的灭菌程序有问题,哈夫金将原本通用的石炭酸灭菌改为
加热灭菌。虽然加热灭菌已在世界领先的巴斯德研究所安全地使用了两年,但英国人并不认可这种方法。
1903年,调查委员会得出结论,擅自更改灭菌程序是疫苗被污染的根源。哈夫金因此被革职,回到了英国。这件事发生在当时法国著名
反犹主义冤案一一德雷福斯事件之后,因此也被称为小德雷福斯事件,原因是它的主角哈夫金和法国军官德雷福斯都是犹太商。后来,李斯
特研究所重新调查了此事,并推翻了判决:他们发现当时一名助手使用了被污染的瓶盖,且未对其进行消毒,因此加热灭菌程序并不是导致
疫苗被污染的原因。1907年,被判无罪的哈夫金回到印度,原本满怀期望准备继续投入工作的他却被禁止测试或生产疫苗,穆尔科瓦尔灾
难就这样在他的生命中留下了不可磨灭的烙印。1914年,哈夫金退休后返回法国,后来移居瑞士洛桑,在那里度过了他生命的最后时光。
姗姗来迟却又全心全意的认可
在威廉·辛普森(William Simpson)爵士的印象中,哈夫金“是一个彬彬有礼、和蔼可亲的绅士,甚至对那些反对他、攻击他的观点和
工作的人也是如此:他非常坚定,能力非凡,充满热情,并且有一种不屈不挠的勇气,不会因失望而却步”。
虽然哈夫金经历了小德雷福斯事件,但他在疫苗学领域所作出的巨大贡献依然获得了公众的认可。1897年,英国维多利亚女王授予哈夫
金爵士勋章,以表彰他在微生物学领域的杰出工作。
1925年,位于孟买的鼠疫研究实验室更名为哈夫金研究所(Haffkine Institute),这也标志着他的声誉在印度逐渐恢复。
当得知实验室更名的消息时,哈夫金回信说:“在孟买工作是我一生中最美好的时光……我祝愿研究所这一国家卫生组织蓬勃发展,并
向全体工作人员致以深深的祝福。”后来,哈夫金研究所成为南亚和东南亚最大的细菌学和流行病学研究中心。
哈夫金于1930年在洛桑去世,享年70岁。犹太电报局发布的一份简短讣告指出,他的鼠疫疫苗“已在印度各地被采用”,他的实验
室“向不同的热带国家发放了数于剂疫苗”。计告还引用了英围细菌学家约夫·李斯特(Josephlister)勋爵的话,他称哈夫金为“人类的数
世主。
20世纪初期,随着“鼠一蚤一人”的传播方式逐渐确立,控制传染源(灭鼠、灭蚤)和改善公共卫生环境等措施有效地遏制了鼠疫大流
行;在印度等地区,哈夫金研制的全细胞灭活疫苗显然也对疫情的控制作出了巨大的贡献。目前,临床上正在开发使用重组毒力因子蛋白的
亚单位鼠疫疫苗,以应对鼠疫作为一种潜在的生物恐怖主义武器的威胁。与灭活的全细胞疫苗相比,亚单位鼠疫疫苗具有快速和高度免疫原
性,并伴有较轻微的局部和全身副作用。虽然灭活的鼠疫疫苗如今已逐渐退出历史舞台,但作为世界上第一支腺鼠疫疫苗的发明者,哈夫金
对抗击传染病的贡献不应被遗忘。
神经科学诺奖史—— 乙酰胆碱的故事
今天的第一位主人公是亨利·哈利特·戴尔(Henry Hallett Dale),他是英国药理学家。在1904年的一天,英国剑桥大学的一个本科生托
马斯·伦顿·埃利奥特(Thomas Renton Elliott)发现在某些情况下,肾上腺素可以模拟交感神经被刺激后的效果。虽然这一发现没有引起人
们的广泛关注,但却吸引了他好朋友戴尔的重视。正巧这一年戴尔在一个药物研究所实习,他就开始研究与肾上腺素存在对结构上相似性的
化学物质的生理学作用,尤其是它们对交感神经的作用。到了1913年,他和小伙伴因为一个巧合,将麦角粗提液直接注射到猫的血液中,结
果发现猫的血压迅速降低了。事后,他们从麦角中提取出了乙酰胆碱。他们相信体内神经纤维可能会释放乙酰胆碱,但受限于当时的技术环
境,他们无法做实验证实。之后战争爆发,戴尔的这些实验就搁置了一阵子。
时间逐渐到了1921年,第二位主人公登场了,他就是德国药理学家奥托·洛维(Otto Loewi)。在一个大早上,他做了一个被人津津乐道
的实验。这个实验非常漂亮,是一个简单的短平快的实验,而且实验设计也颇有来头。洛维后来回忆道:“在1920年的复活节前夜,我醒了。
我打开灯,撕了张纸随便写了点东西,然后又睡了。早上6点,我突然意识到昨天晚上写了很牛逼的东西,但字太丑了认不出来。第二天晚上
三点左右,我又想起来了:这是一个能够验证我17年前提出的关于化学物质传递存在与否的实验设计。我立刻起床奔去实验室,然后根据梦里
的情境做了那个关于青蛙心脏的简单实验。”
在这个著名的实验中,洛维把两个跳动的蛙心分别放进各自的灌流室中,其中一个连有完整的迷走神经,另一个则被去除了迷走神经。随
后,他刺激了连接第一个挖心的迷走神经,使其跳动地更慢;这一现象在当时已经广为人知了。但当洛维将第一个蛙心的灌流液通到第二个
装有第二个蛙心的灌流室中时,神奇的事情发生了:第二个蛙心跳动的速率也大幅度减慢了,和直接刺激第二只蛙心的迷走神经的效果一样
!洛维将灌流液中的抑制性物质命名为“迷走神经素(vagusstoff)”。
后来,戴尔和同事进一步证实了狗的尾部迷走神经可以释放乙酰胆碱。
考虑到他们解决了一个重大的科学问题,即“神经系统的信号传导不仅仅是电传导,而且还是化学传导的”,他们分享了1936年的诺
贝尔生理学与医学奖。
一百年后的今天,我们知道了乙酰胆碱的作用不仅仅是在神经肌肉接头处激活肌肉,在自主神经系统中作为交感神经释放的递质,还是
副交感神经系统中的主要神经递质。我们对乙酰胆碱的产生、降解和重摄取过程也研究地非常透彻了。针对不同乙酰胆碱受体的激动剂、拮
抗剂,我们也应用地相当熟练了。
不过,回顾历史,如果没有戴尔的稳扎稳打的药理学实验,或者没有洛维半夜弯道超车的梦,突触传递的真相将在科学的灰尘中再埋藏
几十年,神经科学领域的发展也可能不似现在这般迅速。
向百年前的科学家致敬。
彩蛋:戴尔的一个女儿后来嫁给了一个化学家亚历山大·托德(Alexander R.Todd),他获得了1957年的诺贝尔化学奖。
访霍乱口服疫苗发明人霍姆格伦:跨越时空 拯救生命
瑞典哥德堡大学微生物与免疫学系教授Jan Holmgren(扬·霍姆格伦)发明了世界上第一个霍乱口服疫苗:Dukoral。目前,Dukoral
是世界卫生组织唯一获准在普通人群中应用的霍乱疫苗,在60多个国家获得应用许可,已拯救众多生命。
鉴于Jan Holmgren在霍乱疫苗研究方面的杰出成果,1994年他获得在欧洲医学领域影响力仅次于诺贝尔奖的Louis Jeantet奖,1977
年获得瑞典皇家学院医学科学奖,2007年国际粘膜免疫学会的卓越科学成就奖等。
近日,他接受《科学时报》专访,谈了成功发明这一疫苗药物的经过及后续研究中的意外惊喜,并表示希望与中国建立合作关系。
霍乱口服疫苗的研制关键
瑞典并非霍乱发生严重国家,Jan Holmgren为何选择远离本国需求的科学问题开展研究?
1969年和1970年在孟加拉国,Jan Holmgren亲眼目睹了令人震撼的霍乱瘟疫。作为科学家,从全球角度做有意义的研究工作是他的
责任和义务。当时,他就下定决心要研究出有效的预防应对办法。同时,瑞典希望帮助发展中国家,因此设立了援外科学基金。
对于研究的成功,他说,这是努力工作加上好运气的结果。
Jan Holmgren说,成功开发出霍乱疫苗的关键,是发现了霍乱毒素B亚基的重要免疫调控功能,另外是采用口服作为给药途径。
其研究表明,口服疫苗比以前常用的注射法更加有效。因为注射法不能在粘膜部位诱发有效免疫,而胃肠、呼吸道和生殖道粘膜表面的
特有免疫系统需要被疫苗刺激后,才能有效激发保护性免疫。因此在各种给药途径中,口服能更好激发肠道粘膜的免疫学反应。这是让霍乱
疫苗有效发挥作用的关键。
疫苗成功经历漫长旅途
一名跟随Jan Holmgren工作了多年的中国学者介绍,早在1884年世界病原细菌学的奠基人和开拓者科赫(Robert Koch)就提出,霍
乱的致病性很可能是由于毒素引起的,但直到上世纪70年代之前,人们还认为毒素是由一个分子构成的。Jan Holmgren的开创性工作纠正
了这一看法。他在1973年首先报道,霍乱弧菌毒素由2个亚基构成——A亚基和B亚基。其毒性主要存在于A亚基,B亚基本身不具备毒性。
自此,Jan Holmgren的团队着重研究了霍乱毒素各亚基的生物学和免疫学特性、粘膜免疫以及粘膜疫苗和给药途径。随后,他尝试了
各种组合来试验霍乱疫苗的效果。他们发现,如果用灭活的霍乱全菌体加上分离出的霍乱毒素B亚基混合做成疫苗,可以经口服产生很好的
粘膜免疫应答,起到预防霍乱感染的作用。因为毒素B亚基能起到很好的粘膜佐剂的作用,辅佐全菌疫苗刺激机体的免疫系统,产生有效的
免疫应答。
他说,霍乱疫苗如同其他疫苗的研发一样,道路漫长而曲折。最初,他们用了5年时间弄清楚机理,首次在《自然》杂志上报道如何构
建霍乱疫苗。此后又经过14年,他们克服了一个个漫漫研发路上的困难,特别是开展了慎重的安全性评估,才把一个概念发展成一个产品,
使得Holmgren的全菌加毒素B亚基的配方,最终成为Dukoral,获得广泛应用。
药品审批本身就是一个漫长的过程,而霍乱疫苗的研发尤其长。因为人体粘膜免疫的机理并不清楚,需要将基础研究、临床免疫学研究
和开发并举。人体的粘膜免疫系统建立了有效的免疫机制,但到目前为止,粘膜抵御外来病菌入侵、避免对外来抗原引起过激免疫反应的机
制并不十分清楚。所以,通过粘膜疫苗来预防感染的研究仍是国际学术界的一个热点。
为了弄清其机理和效果,Jan Holmgren研制霍乱疫苗需要大规模人群试验。为此,他和哥德堡大学微生物与免疫系前主任Ann-Mari Svennerholm
教授合作,指导研究团队在孟加拉国开展了为期5年、9万名志愿者参加的验证。研究过程中,在瑞典援外署的经济援助下,他们帮助孟加
拉国建立了霍乱研究中心。目前,该中心成为世界卫生组织腹泻病合作研究中心。
此外,他们还在莫桑比克和拉丁美洲开展了人群接种试验。
希望与中国建立合作关系
Jan Holmgren说,目前,关于霍乱口服疫苗他们正在进行新一代产品的开发。在近年来和Michael Lebens博士合作开展的后续研究中,
他们获得了一个意外的惊喜,因霍乱毒素B亚基与后来发现的产肠毒素性大肠杆菌(ETEC)结构有相似性,服用Dukoral的接种者,也诱导
出对该大肠杆菌的短期保护力,而ETEC正是导致旅行者出现腹泻的主要原因。
他说,对中国而言,在城市里霍乱听起来似乎是个离人们比较遥远的名词,但中国人口众多,地区广泛,某些地方霍乱并未根除。同时,
与中国毗邻的南亚国家霍乱流行较重,且前往的中国旅游者众多,对霍乱疫苗有一定需求。因此,他希望与中国建立合作关系。
他说,新产品可在一年内进入临床检测。在瑞典可开展Ⅰ期临床试验,因为瑞典人喜欢旅游,招募志愿者并不困难。在第Ⅱ、Ⅲ期临床
试验中,可考虑找更多国家的人测试,以便了解广谱性,包括中国人。因此,中国的任何企业若有兴趣,他都乐意开展合作,进行相关的研
发活动。
霍乱只是腹泻病冰山一角
Jan Holmgren说,霍乱的流行可能被低估了,霍乱只是所有腹泻病“冰山上的一角”。肠道感染引起的腹泻危及儿童和成人的健康,
而仅霍乱全球每年就有300万~500万病例,每年夺去约20万人的生命。
一些霍乱流行的国家,由于缺乏有效诊断技术,不能区别霍乱与普通腹泻;有的地区即使能诊断,但因担心食品出口或旅游业受挫,不
愿意向外界通报其霍乱流行情况。目前,大约一半的腹泻都由与霍乱致病机理相似的细菌引起。
Jan Holmgren介绍,霍乱是所有腹泻病中最严重的。患病者在午饭时完全健康,而在午夜前一命呜呼。当霍乱弧菌侵入体内后,病人
平均每小时会失去2升水。除非及时补充水分和电解质,否则都将致命。
他们的研究表明,很多别的细菌,特别是某些类别的大肠杆菌,也如同霍乱一样通过产生细菌毒素致病,因此霍乱是一个很好的研究此
类细菌腹泻的模型。
Jan Holmgren和中国合作者从霍乱毒素的研究中还发现,其B亚基在某些情况下能促进粘膜免疫耐受,这对治疗自身免疫和过敏类疾
病有重要启发。这是他在研究霍乱多年后的一个意外惊喜。目前,除了继续沿着霍乱疫苗研究这条路径,争取推出新一代性价比更高的产品
外,他还另辟蹊径,开始了基于霍乱毒素B亚基的免疫耐受治疗研究。
Jan Holmgren解释说,一些疾病,比如Ⅰ型糖尿病、多发性硬化、过敏等,往往是由于免疫系统对自身组织或无害异物产生了强烈的
免疫反应,造成了组织伤害。通过进一步研究霍乱毒素B亚基的免疫耐受调控机理,有望开发实用的新型免疫耐受治疗药物。
Jan Holmgren谈起这些研究进展,兴奋不已,充满期待,似乎看到了一个又一个胜利果实。
《科学时报》
避孕药创始人逝世,他的发明推动了女性性权利的解放怎么回事?
“魔丸”的创始人去世了。
6月23日,匈牙利化学家乔治·罗森克兰兹(George Rosenkranz)于家中去世,享年102岁。上世纪50年代,他与同事一起研制出口服避
孕药最关键的成分,他同时也是避孕药的积极倡导者。其子罗伯托·罗森克兰兹(Roberto Rosenkranz)证实父亲的死亡,表示尚不知晓具体死
因。
波伏娃在《第二性》中写:“女性除非获得自由选择生育的权力,否则就不可能真正解放。”而罗森克兰兹带来的这枚药丸,斩断了性
爱与生育的必然联系,赋予女性更大的自由。
1951年,墨西哥城,罗森克兰兹博士和他的同事奥地利科学家卡尔·杰拉西(Carl Djerassi)和墨西哥博士生路易斯·米拉蒙特斯(Luis E. Miramontes)
制造出了炔诺酮。起初,他们是为了研制出防止女性流产的药物,但在这一过程中,他们发现他们正在摆弄的激素也可以防止怀孕。
这是一个令人惊讶的发现,也意味着这枚药丸将面临范围更广的审视——美国正处于婴儿潮中,政治家、宗教领袖、学者等各种角色将
对人类的道德范式展开一场激烈争论。
罗森克兰兹展示避孕药制作工艺
据《洛杉矶时报》报道,他的儿子罗伯托·罗森克兰兹表示,自己的父亲已经充分认识到了避孕药的发现可能对世界、社会规范产生的影
响,但仍旧选择仅仅将其视作“医学进步”。罗伯托说:““他不想参与'与避孕药有关的政治'。” “他认为这是向前迈进的一步。”
最终,炔诺酮成为了避孕药最主要的活性成分。
在炔诺酮诞生后,如何把它制成药、大规模推向市场、并为公众所接受,又是另一桩“不容易”的故事了。许多人都曾为避孕药的面世
贡献出自己的力量,除了乔治·罗森克兰兹和卡尔·杰拉西等人,还有更多的科学家、医生、赞助者、社会运动家参与其中。
《魔丸的诞生》是一本描述避孕药诞生过程的书,英文书名为《The Birth of The Pill》,一语双关——事实上,在避孕药面世时就被
命名为“Pill”,首字母大写以便与药店里的其他药片作为区分,这个专有名词也显示出避孕药的关键地位。这本书在描述其中一位主人公
桑格时,写了这样一段话:
刚开始桑格只是将避孕视为帮助女性控制子女数的方式。而现在,她开始相信,如果能打破性与生育的必然联系,女性将获得她们无法
想象的自由。婚姻将改变,男女关系将改变,家庭含义将改变,女性的就业和教育机会将改变。
关于避孕药最新的消息是,英国的邓迪大学已经开始研发男性避孕药了——据BBC报道,研究人员正用机械臂试验数以千计的化合物,
试图在5年内找出能够阻止精子运动的药物。盖茨基金会捐赠90万美元(约618万元)资助该研究。
能享受最私密的乐趣而不必为生育忧虑,大概是避孕药为女性带来最切实的好处。而以更宏大的视角来看,女性的权益、两性关系的走
向、女性社会角色的转变,都因这枚药丸而变得不同。
来源:网络
新药故事 | 降压药依那普利的成功逆袭
如今,随着国人生活质量的提升,健康日益成为人们关注的焦点。与此同时,由于环境污染、工作压力、饮食不规律等问题,加上现
代医疗检测技术的提高,某些疾病呈年轻化态势,一些从前较为罕见的疾病也变得普遍。于是,开展新药研究就变得极为重要。
从广为人知的降压药,到如今备受关注的HPV疫苗,从价格一度令人瞠目结舌的乙肝疫苗,到有望对抗多种癌症的抗癌药物……,新
药研发的历史也是人类对抗疾病的斗争史。
新药研发的竞争是白热化的,原因之一是,已知可用药的生物靶标是有限的,一经临床前的药理验证,或是更进一步的临床验证,各
大公司的研发资源就都集中在了这些有限的药靶上了。20世纪70年代中后期血管紧张素转换酶(ACE)抑制剂的研发是一个很能说明问题
的实例。
◆血压调控与化学介入
在1628年出版的《心脏与血液的运动》一书中,英国医生威廉·哈维提出了相对完整的血液循环理论,从此人类对于“心血管系统”
的理解进入了一个新时代。进入20世纪,随着血压测量数据的累积,将血压升高作为疾病的描述也日益增多。
血管紧张肽原酶是人类最早发现的蛋白酶,距今已有100多年的历史了。在这100多年里,由血管紧张肽原酶和血管紧张肽所构成的生
物调控体系(RAAS)一直是基础医学和临床研究的热门领域,因为RAAS对人体的体液和电解质平衡以及血压的调控起着决定性的作用。
简单地讲,当人体内血量降低(失血)时,肾脏就会释放血管紧张肽原酶。血管紧张肽原酶将储存于肝脏内的血管紧张肽原通过降解
转变为血管紧张肽-I,新产生的血管紧张肽-I在肺循环过程中被ACE进一步转换为血管紧张肽-II。血管紧张肽-II作为激动剂将其受体激
活,从而引起下游一系列相应的生理变化,最终导致血管壁紧缩,血压升高。这是人体自我保护、保障器官供血的重要机制。如果血压被
维持在较高的状态下,则会增加心梗、脑梗等突发性病变的风险。
值得注意的是,血管舒缓激肽与血管紧张肽一样,主要也是在肺循环的过程中被ACE降解的,由此不难得出结论,ACE对血压调控起
着非常重要的作用,而ACE抑制剂就是20世纪80年代从RAAS体系中研发出来的第一类高效降压药——普利类降压药。
◆卡托普利与药物设计
20世纪70年代初,巴西科学家从美洲洞蛇的毒液中发现了一组多肽,能够增强血管舒缓激肽的功效,被命名为“血管舒缓激肽增强因
子”(BPF)。剑桥大学的进一步研究表明,BPF能抑制从血管紧张肽-I到血管紧张肽-II的转化,正是ACE的抑制剂。
美国施贵宝制药研究团队以这些天然的多肽类ACE抑制剂为起点,采用当时很先进的定位突变生物技术,仔细研究了BPF的构效关
系,他们发现这些多肽C末端的脯氨酸残基对ACE的抑制活性非常重要。以这个脯氨酸残基为核心,施贵宝制药团队通过生物测试发现,
在脯氨酸附近引入巯基能进一步提高化合物对ACE的抑制活性。在巯基和脯氨酸残基这两个结构单元的基础上,他们找到了高效率的ACE
抑制剂,成功地设计出了卡托普利。
1981年4月,施贵宝制药将第一个普利类降压药卡托普利推上了市场,开启了RAAS体系药物研发的新时代。这是第一个以化学结构
为基础的新药设计的成功例子,从那以后,这种先进的思想方法得到了广大药物化学家的接受,成为现代药物化学的主流。
◆伊纳普利与更优药物
面对施贵宝在ACE抑制剂研发方面的领先局面,美国默沙东公司的研究人员一直密切地关注着卡托普利的研发。默沙东研究人员基于
从其他项目得来的类似经验,认为巯基很有可能造成三种类型的副作用:白细胞降低、皮疹和影响味觉(后来的临床研究报告证实了这一
推断)。于是,默沙东的课题组把设计不含巯基的ACE抑制剂作为新的目标。
为了赶超卡托普利,默沙东的团队应用了“前体药”的概念,将羧基转化为乙酯,成功地研发出了第二个在美国被批准使用的ACE抑
制剂——依那普利。
依那普利本身的活性并不高,必须经肝脏的酯酶水解后才能产生高活性的二羧酸依那普利,即依那普利拉。依那普利口服后吸收迅
速,生物利用度约60%(不受食物影响),虽然依那普利在1小时内达到血浆峰值浓度,但依那普利拉则需3~4小时才能达到血浆峰值浓
度。依那普利拉与ACE的结合非常紧密,因此血浆半衰期约为11小时,非常适合每日一次的服药间隔。除了普利类药物共有的一些轻微的
副作用(如干咳)外,依那普利即使在高剂量服用时也没有卡托普利特有的副作用。
由于上述优点,依那普利在1985年12月被批准上市以后,销售额迅速而又稳步地上升。
在继续深入研究RAAS血压调控体系的过程中,默沙东又相继研发出疗效更好、更安全的更优专利药。在1995年把新一代的降压药氯
沙坦(商品名科素亚)推上了市场,而海捷亚则是科素亚与利尿型降压药氢氯噻嗪的复方制剂,双管齐下,效果更佳。
本文转载自读者报 编辑:何建 责任编辑:董小玥 审核:周华
最美丽的毒药——肉毒素的发现史
腊肠是古今中外人们都非常喜爱的一种腌制食品,但如制作方法不当,也偶尔会导致人死亡。1820年的某一天,德国的某个小镇上,有
居民在食用腊肠后发生了头痛、乏力、恶心、呕吐等症状,甚至有人肌肉麻痹、肢体瘫痪。一位叫克尔纳(Justinus Kerner)的医生对此次
中毒事故患者的症状做了详细的记录,并决定把这种当时还未被人发现的致毒物质称为“腊肠毒素”、“肉毒之毒”(来源于拉丁文
“botulus”——火腿)。这是首次对食物源性肉毒中毒的临床症状进行准确和完整的描述。同时,克尔纳医生还通过在动物和他自己身上做
实验发现,这种毒素是在厌氧条件下产生的,能够在不影响感官和精神功能的情况下,阻断躯干和自主运动,如果被大量摄入是可以很快致命
的。类似的事件在1895年又再次重演。比利时又出现一起因吃腌制火腿而导致的中毒事故,34个出席葬礼的人因吃了没有充分腌制的火腿,
纷纷中毒,症状同克尔纳医生以前记述的肉毒中毒的麻痹症状一模一样。比利时细菌学家Emile Pierre Van Ermengem教授从尸体和食物中
分离出了一种细菌——肉毒梭状芽孢杆菌。这种细菌是导致受害者全身性肌肉麻痹的原因,这些受害者死于胸部肌肉麻痹后的窒息。
多数情况下,这类食物在食用前都经过充分的烹饪,所以肉毒杆菌的食物中毒事件,其实是相当偶然的。直到应军事的需求,推动了罐头
类食品的诞生和大规模应用,肉毒杆菌食物中毒事件,在人类史上才进入高发期。事实上,正是由于美国历史上糟糕的罐头、火腿、香肠生
产商,制造了太多食物中毒事件,才因势利导推动了如今声名赫赫的美国FDA(食品和药物管理局)的诞生。这些中毒事件中最知名的受害
者就是美国总统西奥多·罗斯福。1898年,老罗斯福曾带领一支志愿骑兵队去古巴参加美西战争,悲剧的是,罐头导致的食物中毒,让他部
队非战斗减员就达数千人之多,其中上百人因此死亡。当老罗斯福成为美国总统后,一本揭露美国食品工业内幕的流行书籍《丛林》,让他
下定决心推动相应食品安全立法,并在1906年获得成功。但这直接影响了罐头食品业的发展。直到1920年,一名瑞士裔美国兽医科学家
Karl Friedrich Meyer发现了加热法防止肉毒素中毒的方法,才挽救了当时岌岌可危的罐头食品业。
正如黑格尔的名言一样,“存在即合理”。即便是毒性如此之高的肉毒毒素也并非一无是处。20世纪20年代以后,对肉毒毒素的研究逐渐
走出了一条新路。
首次纯化A型肉毒毒素的尝试是在1928年由Herman Sommer在美国旧金山加利福尼亚大学Hooper基金会开展的。Sommer博士从用
过的神经毒素微生物的培养液中以高浓缩的方式沉淀了A型肉毒毒素。1949年,英国剑桥大学的Arnold Burgen博士团队发现了肉毒素作用
机理:肌肉运动离不开神经末梢释放乙酰胆碱,而肉毒素阻断神经末梢乙酰胆碱的释放,从而导致运动神经与自主神经瘫痪,麻痹胆碱能支
配区肌肉和骨骼肌。但同时,毒素也可能有助于降低活动过度的肌肉的活动水平。
受此启发,肉毒素被用于斜视的治疗。当时专注治疗斜视(“斗鸡眼”)的眼科医生已经可以利用肌动电流图来找到具体是哪一块肌肉
导致了斜视。因为传统治疗斜视的眼科手术风险高而且需要多次重复治疗,眼科医生们一直在尝试运用注射的方式进行治疗,他们先后尝试
了:各种麻醉剂、酒精、各种酶、各种拮抗剂,最后还用上了蛇毒,但效果都不理想。最后,受到约翰霍普金斯大学Daniel Drachman用
肉毒素给鸡注射的实验结果启发,Alan B Scott博士和他的同事将A型肉毒素注射到猴子身上,取得了惊人的效果:只需要几皮克(10 -12
克)的肉毒素就能够不扩散地麻痹目标肌肉,而且持续时间长,没有其他副作用。随后Scott团队用全力投入A型肉毒素制剂的研发,他们解
决了把肉毒素做成可以药用的制剂需要解决的一系列技术问题:冻干、白蛋白缓冲溶液的配制、消毒、确认效价和安全性,现在肉毒素的制
备工艺虽然有很大的改进,但也基本是这个流程。
临床前动物研究显示有前景后,Scott博士于1970年代后期获得了FDA在人体上研究A型肉毒毒素用于斜视的许可。1982年,Scott博士
和小部分医生已经开始开始用A型肉毒素对这些疾病进行小范围治疗。此后,他还进行了第一例斜颈的治疗。渐渐地,在一些特殊病人群里
流传开了“A型肉毒素能够治疗不治之症”的传闻。之前无法治疗的运动功能障碍,比如导致功能性失明的眼睑痉挛在美国各地都慢慢开始
出现成功治愈的例子。而斜颈病人也发现,尽管震颤无法解决,他们的疼痛能够被肉毒素注射极大地缓解,运动能力也得到了加强。病人的
需求和呼声推动了更多的神经科医生、整形科医生或康复科医生开始了小心翼翼的尝试。他们慢慢尝试用肉毒素治疗中风后肌肉痉挛、肌张
力失常、和斜颈相关的肌肉挛缩,对待肉毒素的眼光也开始从不信任变得比较理性。1989年, 美国FDA批准A型肉毒毒素(商品名BOTOX作
为第一个微生物毒素临床药物上市 。
中国治疗用A型肉毒毒素(衡力,BTXA)的开发研究始于20世纪80年代中期,略晚于美国。由于兰州生物制品研究所有国家梭菌实验室的
基础,积累了不少肉毒梭菌培养、肉毒毒素精制提纯方面的学识和经验,使治疗用A型肉毒毒素很快完成了实验室研制。在取得试制品治疗
斜视、眼睑痉挛、面肌痉挛等I、Ⅱ期临床试验安全、有效结论的基础上,1993年10月经中国卫生部批准为新药,并获得试生产文号,
1997年2月转为正式生产文号。从此,该药在全国31个省、市、自治区乃至台湾和香港推广使用。
2002年4月15日美国FDA正式批准BOTOX的美容适应证,肉毒素成为了“最美丽的毒药”,一场肉毒毒素应用热已席卷全球。但国内、
外有关专家不无担心,美国FDA批准BOTOX美容用途后,缘于人们对美丽容颜的期盼和巨大商机的追求,肉毒毒素的生产、供应将大幅度地
增加,从而带来诸如公众健康和国家安全受到威胁的更多麻烦和危险。
本文转载自搜狐网--医学的历史与文化
“药王“修美乐诞生记和专利战
医药云端工作室:挖掘趋势中的价值
来源:药事纵横 作者:二友木
前 言
2002年全球第一个全人源单克隆抗体阿达木单抗(商品名:Humira/修美乐)获美国FDA批准上市。自2012年始,Humira已经连续
七年拿下全球处方药销量第一的宝座,并在2018年达到了199.36亿美元的全球销售额。
迄今为止,Humira全球累计销售超过 1000 亿美元,是当之无愧的“药王”!。阿达木单抗是继英夫利西单抗和依那西普之后,获得
美国FDA批准的第三个TNF抑制剂类药物。
修美乐诞生记
上世纪80至90年代,实验模型和类风湿关节炎患者的研究阐明了细胞因子在类风湿关节炎中的作用:滑膜中巨噬细胞和成纤维细胞过
度产生促炎细胞因子似乎是导致慢性炎症的关键因素,这些促炎细胞因子主要包括肿瘤坏死因子(TNF)-α,白细胞介素(IL)-1和IL-6。
细胞因子级联反应导致类风湿关节炎:活化的T细胞侵入滑膜并释放细胞因子促使滑膜巨噬细胞和成纤维细胞产生炎性细胞因子
(TNF-α,IL-1和IL-6),从而导致复杂的信号转导级联和产生参与组织降解的分子以及导致骨的再吸收和破坏的分子。
1989年,英国的Brennan等开展了一项颇具里程碑式意义的实验。在该实验中,他们对类风湿关节炎(RA)滑膜组织培养物产生的细胞因
子进行了阻断。
结果发现,对肿瘤坏死因子(TNF)-α进行阻断后,可下调白细胞介素(IL)-1、粒细胞-巨噬细胞集落刺激因子(GM-CSF)、IL-6和IL-8,以及
其他所有被检测的促炎细胞因子、半数以上的趋化因子和其他多种活性分子,如基质金属蛋白酶(MMP)等。
这些试验结果支持“TNF-α是RA的关键致病因子,同时也是RA的重要治疗靶点”这一论点。最先获批应用于临床的抗TNF-α制剂包
括英夫利昔单抗、依那西普和阿达木单抗,其中阿达木单抗的表现备受瞩目,是全球首个获批的全人源单克隆抗体。
阿达木单抗是重组人源IgG1单克隆抗体,含有人源轻链和重链可变区序列和人源IgG1;英夫利昔单抗人鼠嵌合IgG1单克隆抗体(由人
源IgG1恒定区和小鼠可变区组成)
阿达木单抗最初由CambridgeAntibody Technologies(CAT)和BASF子公司BASF Knoll合作开发。2002年,雅培制药公司
(Abbott)以69亿美元收购BASFKnoll而获得阿达木单抗。
BASF Knoll在之前与Biogen的合作虽然没有把TNF制剂成功地商业化,但是从合作中吸取了丰富的TNF制剂开发经验。当时,Knoll
还开发了抗TNF小鼠单克隆抗体MAK195用于急性反应,如败血症(afelimomab,Segard)。
MAK195虽然能强有效地抑制TNF,但是作为鼠源抗体,MAK195并不被考虑用于治疗慢性自身免疫疾病,因此该公司决定开发一种
具有低免疫原性和长血浆半衰期的新型人源抗体。
以MAK195作为模板,采用CAT公司的抗体噬菌体展示技术,引导选择人抗体V结构域,从而达到与TNF相似表位的高亲和力结合的
目的。分别构建两个单链抗体库,其中一个库具有MAK195VH,结合人类可变轻链库,另一个库具有MAK195VL,结合人类可变重链
库。
两个库用来筛选与TNF结合的抗体,将筛选出来的VH和VL重新组合后成新的库,继续用来筛选TNF结合的抗体。随后进行CDR突
变,D2E7(阿达木单抗)就诞生了。
Knoll完成了临床候选药物D2E7的大部分药物开发过程,随后Knoll被出售给Abbott公司,并由Abbott对D2E7进行进一步开发和营
销。在临床试验中,阿达木单抗具有长期疗效和安全性,即用型液体制剂和每隔一周给药,可选择与甲氨蝶呤(MTX)联合使用。
阿达木单抗于2002年12月31日首次获FDA批准用于治疗中度至重度形式的风湿性关节炎,作为单一疗法或与MTX或其他改善疾病的
抗风湿药物联合治疗,随后由Abbott公司以“Humira”商品名进行销售,该商品名代表了“human monoclonalantibody in
rheumatoid arthritis”。
阿达木单抗是继嵌合单克隆抗体英夫利昔单抗和TNF受体融合蛋白依那西普之后的第三个获得批上市的TNF抑制剂。尽管市场起步较
晚,但阿达木单抗已发展成为全球最畅销的抗体药物,2013年全球销售额超过100亿美元。与英夫利昔单抗(每4-8周静脉注射3-10
mg/kg)相比,阿达木单抗每2周皮下给药40 mg/kg,停药风险更低,给药更简单(自动注射针)。
目前为止,阿达木单抗在美国、欧盟和日本获批用于成人中重度慢性类风湿性关节炎和儿童多关节少年特发性关节炎、银屑病和银屑
病关节炎、强直性脊柱炎、儿童和成人克罗恩病、溃疡性结肠炎(仅限美国和欧盟)等,目前正在进行结节病和葡萄膜炎治疗的临床试
验。阿达木单抗目前在中国仅获批3个适应症,分别为类风湿性关节炎、强直性脊柱炎和斑块状银屑病。
阿达木单抗的治疗紧急不良反应包括结核病等潜伏感染的再激活,或更高几率的感染患病率,主要原因是由于TNF被抑制。
根据Humira注射处方信息和药物指南,淋巴瘤的风险比普通人群高3倍,但与类风湿性关节炎患者相比,肿瘤风险与抗肿瘤坏死因子
治疗无显著差异。
阿达木单抗适用于已对英夫利昔单抗不耐受的成人克罗恩病患者,而不适用于抗肿瘤坏死因子阻滞剂耐药的溃疡性结肠炎患者。
修美乐首篇专利概况
阿达木单抗(当时称为D2E7)的专利申请于1996年首次提交,首次发布为WO9729131A1。Eisai于1999年获得阿达木单抗在日本、
韩国和台湾的共同开发权。
雅培于2000年以69亿美元收购了Knoll,随后在2013年分拆为两家公司,AbbVie成立为研究型制药公司。CAT公司持有的阿达木单
抗专利权于2006年10月由阿斯利康出售给了Royalty制药公司。
尽管这些专利是等效的,但其申请数量不同,两项专利(US6090382和EP0929578B)所包含的权利要求均少于原PCT申请。美国和
欧洲专利的前20项权利要求是相同的,但美国专利仅包含10项从属权利要求。相比之下,欧洲专利包含27项附加权利要求,其中包括对
分离核酸的权利要求,以及各种使用方法的权利要求。
AbbVie的专利战略
2014年,Zydus CadilaHealthcare Ltd.在印度推出了第一款阿达木单抗生物仿制药(Exemptia),价格仅为Humira价格的五分之
一。
阿达木单抗获得许多专利保护,修美乐受益于美国和欧洲的专利延长,在欧洲(国家)已获得补充保护证书,其有效期延长431天直
至2018年10月16日。
在美国,US6090382的期限延长326天至2016年12月31日。然而,在讨论2015年第三季度业绩时,AbbVie表示,预计其专利组合
中的70多项专利将阻止美国在2022年之前推出阿达木单抗生物仿制药。
Abbvie开发的大型专利组合是由一个原始申请中衍生出多个分区专利的结果,包括72项授权专利和另外26项专利申请组成的专利组
合。
因此,11项专利的标题都是“用于治疗肿瘤坏死因子-α相关疾病的人类抗体的配方”,所有这些都来源于2002年提出的原始申请。
药物简史│氯吡格雷:基于表型筛选的药物发现
氯吡格雷(Clopidogrel)是一种噻吩并吡啶分子,在结构上与噻氯匹定(Ticlopidine)相关,后者是此类抗血小板药物的第一个成员。
故事始于 1972年,Jean-Pierre Maffrand的经理Fernand Eloy博士决定寻找替诺立定(Tinoridine)相关的新型抗炎药,这是一种噻吩并吡
啶化合物,其抗炎和镇痛特性已于2年前由Yoshitomi公司发表。
利用在噻吩并吡啶化学方面的知识,Maffrand等合成了许多克级衍生物,随后利用动物模型对这些化合物进行筛选。大部分的测试都是
在小鼠和大鼠体内或体外进行的。结果显示,合成的噻吩并吡啶化合物似乎没有一种具有抗炎或镇痛作用,但是,其中一些化合物在对大鼠口
服给药后表现出意想不到的抗血小板和抗血栓形成活性。值得一提的是,当时寻找新的抗血小板药物并不常见。血小板聚集、血栓形成与心血
管事件之间的联系仍被一些心脏病专家忽视或处于争论之中,事实上,血管痉挛被认为是动脉粥样硬化临床并发症的主要原因。发现的活性
最好的化合物之一噻氯匹定被迅速选择用于开发,并首先在血小板与人造表面相互作用可导致血栓并发症的临床条件下进行评估,例如在体外
循环心脏手术或接受血液透析的患者中。
噻氯匹定于1978年在法国上市,名称为Ticlid。随后的大型临床试验证明了噻氯匹定在其他血栓形成事件高风险患者中也有效,特别是
那些既往有短暂性脑缺血发作和中风、外周动脉疾病或缺血性心脏病的患者。该产品随后在全球范围内使用,并于1991年进入美国市场。
噻氯匹定进行临床前开发的同期,Maffrand等继续合成噻吩并吡啶类似物,以尝试获得在动物中具有更好活性/毒性比的备用化合物,
这可以转化为更好的人类获益/风险比。在法国推出噻氯匹定几个月后,这一目标变得更加重要,因为一些接受治疗的患者似乎患有严重的血
液系统疾病,包括白细胞减少症、血小板减少症、粒细胞缺乏症和全血细胞减少症。这些危及生命的不良反应在随后的大型临床试验和上市
后药物警戒中得到证实和量化。它们中的大多数发生在治疗的前3个月,并且在停止治疗时可逆,但不幸的是,它们无法通过任何已识别的
人口统计学或临床特征进行预测。因此,卫生当局要求在这3个月期间必须对接受噻氯匹定治疗的患者进行血液学和临床监测。
总共合成了一千多种噻氯匹定类似物,并在动物身上测试了它们的抗血小板和抗血栓形成作用。其中八个已在健康志愿者中进行到I期
研究,只有PCR4099被证明比噻氯匹定更有效,耐受性更好。进一步的初步II 期临床研究证实了其对先前发生血栓事件的患者具有强大的
抗血小板作用。
PCR4099是等量的两种对映异构体的外消旋混合物。由于担心噻氯匹定在人体中的不良反应,所以Maffrand等决定尝试分离两种对映
异构体,看看其中一个异构体是否可能具有更好的活性/毒性比。研究表明,只有右旋(S)-异构体(氯吡格雷)具有抗血小板和抗血栓形成
作用,而无活性的 (-) - (R)-异构体显然在动物中耐受性较差。因此,外消旋 PCR4099 的临床研究于1987 年停止,同时开始了硫酸氢氯吡
格雷的临床前开发。 经过10年的开发和大规模临床研究,硫酸氢氯吡格雷于1998年在全球范围内推出。
当氯吡格雷进入临床前开发阶段时,其作用方式(以及噻氯匹定的作用方式)尚未完全阐明,但明显不同于其他血小板抑制剂,如阿司
匹林、磺吡酮和双嘧达莫。与这些药物不同,它似乎是ADP诱导的血小板聚集的强效抑制剂。由于不同作用机制,氯吡格雷和阿司匹林的组
合增强了它们在动物中的抗血小板和抗血栓形成活性。此外,氯吡格雷是一种前药:它在体外没有活性,需要肝脏代谢才能有效。我们确信
活性代谢物在化学和生物学上是不稳定的,并且它在血小板的剩余生命周期中不可逆转地影响血小板,从而解释了其作用持续时间长。
氯吡格雷活性代谢物于2000年被分离和表征,其血小板靶点于2001 年被鉴定为ADP的P2Y12受体。参与氯吡格雷活性代谢的酶被逐步
鉴定,最后一种 (PON-1) 于2011年提出,但随后未得到证实。也就是说,从发现噻氯匹定后的30多年和合成氯吡格雷后的10多年,才阐
明了这两种主要抗血栓药物的作用机制。
氯吡格雷由赛诺菲和百时美施贵宝以商品名Plavix销售。 2010年,它以94亿美元的全球销售额位居第二,但仿制药现已上市。第三代
噻吩并吡啶类药物普拉格雷最近被批准用于减少接受经皮冠状动脉介入治疗(PCI) 的急性冠状动脉综合征患者的血栓性心血管事件(包括
支架内血栓形成)。
最后,至少可以从噻氯匹定和氯吡格雷的故事中吸取两个教训。首先,应该更多地关注现在使用全细胞的表型筛选,以补充更容易执行
的基于目标的筛选。其次,不应因为遇到某些最终可以控制的缺点而过快地放弃候选药物。如今药物发现主要依赖于基于靶点的筛选,那么
今天我们是否还能发现和开发向氯吡格雷这样的药物呢?
参考文献
Maffrand, Jean-Pierre. "The story of clopidogrel and its predecessor, ticlopidine: Could these major antiplatelet and antithrombotic
drugs be discovered and developed today?." Comptes Rendus Chimie 15, no. 8 (2012): 737-743.
本文转载自知乎:青梅煮药 香港理工大学 哲学博士
人血白蛋白的“前世今生”
小编寄语
“生存,还是毁灭?这是一个问题。”如今,对许多晚期肿瘤患者来说,这个“问题”集中在 一个小小药瓶上,而药瓶里装的是几毫升乃
至几十毫升的药水——人血白蛋白,临床上一直 称其为癌症病人晚期维持治疗的“救命药”,今天小编就带领大家来一睹人血白蛋白的“前
世今生”。
你绝对想不到 |人血白蛋白的诞生完全是一个“政治任务” 在第二次世界大战期间,生灵遭到涂炭,对安全稳定的血浆替代品的需求急剧
增加。
此时,哈佛大学医学院的Cohn教授与同事发明了低温乙醇分离法来纯化白蛋白,该方法作为 血液制品生产经典工艺沿用至今。
然而,故事远远没有看上去那么简单…… 1940年美国输血委员会批准白蛋白可作为血浆替代物用于临床。不过,在1940年由于原料来源
以及产量的考虑,不是生产的人血白蛋白,而是牛血白蛋白!牛血白蛋白输入人体后可想而 知会发生什么情况,当时至少有两人死于急性肾
衰。
可惜的是开始的时候人们以为是由于白蛋白不够纯,有杂质的原因,于是忙于改进方法,达到 了纯品中球蛋白仅占0.008%的程度,可是
疗效仍然未出现,突然有一天,有人不经意的问了 一句:是不是因为物种不对啊?直到这时医学家们才想到是物种的问题,并立即转向为人
血白 蛋白的提取和应用。
人血白蛋白的纯化提取工作开始于1941年,血液原料来自于美国红十字会。当年的美国军医 I.S. Ravdin将当时能得到的全部人血白蛋白
用于1941年12月7日珍珠港轰炸中受伤的7个严重 烧伤与休克的士兵,士兵均存活!
随后该疗法被迅速推广。之后美国7个商业试验室参与纯化白蛋白的工作,总计利用了2百万 单位的人血生产了近600,000 12.5-g单位
的人血白蛋白。在唯一的临床观察研究中,185例志愿者没有发现任何不良反应。
人血白蛋白全拜二次世界大战荼毒生灵之赐来到人间,而且临床应用前几乎没有进行相关的病 例对照研究,完全是战争的原因让这个药
物自己证明了自己。
本文来源:生物制品圈
闲聊“片仔癀”
片仔癀是蜚声中外的传统中成药,具有清热解毒,凉血化瘀,消肿止痛等功效,越来越受到医药专家和患者的青睐。
片仔癀名字的由来
福建漳州方言把热毒肿痛统称为“癀”,而片仔癀的外观为类扁椭圆形块状,既可内服又可外敷。因一片即可退癀,故名“片仔癀”
(“仔”为福建闽南方言中的语气词)。
片仔癀曾是宫廷秘方
片仔癀的面世可追溯到距今近500年前的明朝,那时的片仔癀可是明朝太医院的“御用良药”。明朝嘉靖年间,一位御医因不满朝廷暴
政,携秘方逃离皇宫,几经辗转来到福建漳州,隐姓埋名,最后在漳州东郊的璞山岩寺削发为僧。寺庙中多有习武之人,经常舞刀弄枪,身
伤骨损在所难免。这位御医出身的寺僧根据带出的宫廷秘方制成药锭,为习武的寺僧接骨疗伤,疗效显著。
片仔癀是璞山岩寺镇寺之宝
据说这位御医出身的寺僧凭宫廷秘方制成的药锭含有麝香、牛黄、蛇胆、三七等中药材。寺庙附近的百姓若有跌打损伤,寺僧也广为救
治,由于在疗伤时只需用刀切下一小片,故民间将其称为“片仔癀”。渐渐此药在社会上享有崇高的声誉,被誉为“佛门圣药”。寺僧临终
前,将秘方和制作工艺传授给住持,并嘱咐,此秘方传内不传外。从此,片仔癀在寺内代代相传,秘不外泄,成为璞山岩寺的“镇寺之宝”。
片仔癀秘方传至民间
清末,片仔癀的秘方传到了寺僧修文的手中。由于当时求药的人太多,加之片仔癀所用原料均为珍贵药材,价格昂贵,修文为生计所迫
,不得不改送为卖,将片仔癀制成成品,寄托在东门外的“馨苑茶庄”售卖。
修文圆寂后,片仔癀秘方和制作技术由其弟子延侯继承。后来,社会动荡,璞山岩寺日渐式微,延侯还俗,并娶“馨苑茶庄”的李珠为
妻,继续在漳州东门外的馨苑茶庄制售“僧帽牌”片仔癀。由此,片仔癀由佛门传入民间,并被闽南旧时风俗奉之为“镇宅之宝”。当地人
拜访长辈亲戚素有送片仔癀的习惯,并随着华侨流入南洋,赞誉如潮,在东南亚享有崇高地位。
片仔癀成为国宝名药
新中国建立后,馨苑茶庄划归医药行业。1956年,政府对个私企业进行改造,漳州“馨苑茶庄”与“同善堂”“天益寿”等9家药店合
并成立公私合营同善堂联合制药厂,1957年12月,同善堂联合制药厂与公私合营存恒联合神粬厂合并,改名为公私合营漳州制药厂,片仔
癀为漳州制药厂主导产品。1966年漳州制药厂由公私合营转为地方国营,停用“僧帽”牌商标,改用“荔枝”牌商标,片仔癀的生产走上
了新的发展道路。至1990年,片仔癀已经成为福建省出口创汇最多的中成药。1992年,片仔癀处方和工艺被列入国家秘密,成为国宝名药。
经过片仔癀人几十年的创业,企业由小变大,由弱变强,逐渐发展壮大。1993年,以漳州市制药厂为核心企业成立漳州片仔癀集团公司。
1999年12月,以漳州片仔癀集团公司为主要发起人,联合其他法人单位共同发起设立了漳州片仔癀药业股份有限公司。
片仔癀是由漳州片仔癀药业股份有限公司独家生产的传统名贵中成药,其处方、工艺均属国家绝密级秘密,是国家一级中药保护品种。
请大家注意,片仔癀目前只有一个生产厂家哟!
片仔癀的处方虽是国家绝密级秘密,但我们仍可从已公开的部分药物组成中了解到这一名贵中成药的基本功效。已公开的药物组成是天
然牛黄、天然麝香、三七、蛇胆。牛黄可清心解毒,豁痰开窍,凉肝息风;麝香可开窍醒神,活血通经,消肿止痛;三七可散瘀止血,消肿
定痛;蛇胆可清肺化痰,消热解毒,清肝明目,利胆。这四味药与其他药物配合,使得片仔癀具有清热解毒,凉血化瘀,消肿止痛的显著功
效。现代研究表明,片仔癀具有抗肿瘤、保肝、清热解毒、抗炎等方面的药理作用,临床应用十分广泛。在此小编就把片仔癀的临床应用罗
列如下。
治疗肿瘤
临床数据表明,片仔癀可抑制癌细胞的增殖,具有较强的免疫增强作用,可显著提高肝癌、结肠癌、食道癌、骨肉瘤、卵巢癌等患者的
生活质量,减轻化疗副作用,延缓患者病情进展。
治疗肝炎
片仔癀具有保肝、降酶、促进肝细胞修复再生的作用,所以,可用于急慢性病毒性肝炎患者及黄疽型肝炎患者的治疗,临床痊愈率、显
效率均明显提高。临床实验表明,片仔癀治疗病毒性肝炎的临床疗效优于常规西药治疗,且对急性病毒性肝炎的治疗效果好于对慢性病毒性
肝炎的治疗。
治疗口腔溃疡
片仔癀治疗复发性口腔溃疡或放射性口腔溃疡具有很好的疗效,效果优于锡类散,但价格也很美丽!
治疗静脉炎
对于化疗性静脉炎,可将片仔癀粉直接湿敷于病变处,无需纱布等外物,其止痛效果明显,消肿快,起效迅速,舒适度好,方法简单,
疗程短,患者易接受,且效果优于硫酸镁湿敷的治疗效果。
治疗无名肿毒、无名高热等
片仔癀具有一定的解热抗炎作用,其作用与阿司匹林相当。所以,对痈疽疔疮,褥疮,无名肿毒,莫名高热,跌打损伤及各种炎症均有
良好疗效。
片仔癀还具有脑保护和促进乙醇代谢等作用。随着对其研究的进一步深入,片仔癀的应用范围会不断扩大,会有更多的患者受益于片仔
癀。毒理实验研究结果表明,片仔癀具有较好的安全窗口,常规剂量服用片仔癀是安全的。
但因片仔癀含有麝香,故孕妇及经期女性慎用。此外,作为消炎解毒的圣药,片仔癀还不适用于虚症,例如气虚,阳虚,阴虚等,湿热
湿寒的患者也不能用。所以,片仔癀虽有较好的安全窗口,但到目前为止,片仔癀仍属于治病的药品,不可随意作为保健品使用!
本文转载自中医药博物馆
(北中医博物馆 卢颖)
三十年磨一剑,奥美拉唑的“发家史”
“十人九胃”,很多人都有胃病,消化科也是人满为患。事实上胃酸过多是胃肠道疾病,如反流性食管炎的病因之一,而奥美拉唑作为
第一代的质子泵抑制剂(PPI),可以有效且持久地抑制胃酸分泌,从而缓解胃部的不适,属于消化科较为常用的药物之一。
19世纪20年代,一个叫圣马丁的美国男子胃部受伤,战地医生博蒙特赶到现场为他进行了救治。圣马丁被救活了,但是胃部却留下了
永久的伤口。非常有心的博蒙特医生将他带回了家,采取将食物倒进其胃内的方法来观察整个的消化过程,从而发现了游离的胃酸。
事实上胃酸具有帮助胃肠道消化蛋白、吸收钙、铁离子及帮助人体抵御胃肠道细菌感染等作用,但是过多的胃酸分泌则可能会导致反流
性食管炎,临床表现为烧心、嗳气、消化道溃疡等。
在三十年之前,消化道溃疡对于千百万世人来说是一种威胁生命的严重疾病,患者饱受该病的痛苦与折磨。
对于胃酸过多的治疗当时主要是通过服用抗酸药来中和胃酸,这种方法治标不治本,只是能暂时缓解病情,不可能治愈。
到了上世纪70年代,葛兰素史克公司上市了一种组胺受体(H2)阻滞剂西咪替丁。西咪替丁与后来作用机制相类似的药物如雷尼替丁、
法莫替丁等可显著地抑制胃酸分泌,是成千上万胃酸过多患者的“大救星”,这类药物能显著改善病情,提高患者的生活质量。
然而正当西咪替丁、雷尼替丁为争夺治疗胃溃疡霸主的地位斗得热火朝天的时候,一个新型的质子泵抑制剂类药物——奥美拉唑开始慢
慢地对它们产生了威胁。这是因为H2受体阻滞剂可能会受到食物的刺激而不能完全地抑制胃酸分泌,而奥美拉唑对于胃酸分泌的抑制作用则
与刺激方式无关。
奥美拉唑
这个药物的研发可谓命运多舛,真的是三十年磨一剑。
1、一开始因为有人发现麻醉剂可以抑制胃酸分泌,在60年代初阿斯利康公司开展了一项胃肠研究计划,意图通过麻醉剂的类似合成物
来抑制胃酸分泌。该项研究以利多卡因为基础,合成了多种相关化合物。
2、在1970年合成了对大鼠胃酸抑制有很好效果,但是对人体无效的化合物H81/75,使得该项研究几近搁浅。
3、到了1972年,抑制胃酸的项目以新的方法重新被启动,通过文献检索,科研工作者发现了施维雅公司开发了一个抑制胃酸分泌的化
合物CMN131,缺点是存在急性毒性,但事后证实该化合物中的硫代酰胺是引起毒性的主要原因,那么对其进行结构的修饰掩盖是否就可
以降低其毒性呢?
4、终于,在1973年科学家们发现了较为安全的苯并咪唑H124/26。但是不幸的是该化合物已被其它公司的专利覆盖,最终以H124/26
的代谢物H83/69(替莫拉唑)作为先导化合物进行进一步研究。然而,研究又发现该化合物存在着引起甲状腺肿大及胸腺萎缩的长期慢性
毒性,因而对H83/69再次进行结构修饰势在必行,由此得到了化合物H149/94(吡考拉唑)。为了提高药物的膜穿透力、在酸性部位的聚
集能力,科学家对H149/94再次进行了结构修饰,最终获得了奥美拉唑。
5、新药申请在1980年提交,1982年进入实质性实验阶段。
然而,奥美拉唑在大鼠的终生大剂量毒性实验中,发现大鼠体内出现内分泌腺增生与肿瘤的严重问题,导致在1984年中止了该药所有的
临床研究。但不久,在高剂量给与H2受体阻滞剂或者产生高胃泌素血症的外科手术的的研究中也发生了内分泌肿瘤的病例,这使得对于奥美
拉唑的研究得以重新开始。之后涉及13个国家的45个医学中心的多中心研究证实,奥美拉唑使用后,95%的胃溃疡和十二指肠溃疡可在6周
内被治愈,胃及十二指肠溃疡的手术数量大幅度减少,结果表明奥美拉唑在治疗胃溃疡上的效果要优于雷尼替丁。
但在奥美拉唑上市后的再评价过程中发现了患者个体间存在着较大的差异,很多患者需要更长疗程、更大剂量来进行治疗。1987年,阿
斯利康又启动了新的质子泵抑制剂的研发计划,最终发现奥美拉唑的光学异构体(S型异构体)埃索美拉唑,并在2000年以商品名
Nexium(耐信)上市。
这就是抑酸“神药”奥美拉唑的“发家史”,可谓三十年磨一剑。
虽然该药存在个体差异,但仍是消化科常用的质子泵抑制剂之一,且引领了一大批质子泵抑制剂先后问世。目前该类药物主要用于十二
指肠溃疡和卓-艾综合征的治疗,也可用于胃溃疡和反流性食管炎的治疗,此外与某些抗菌药物合用还可用于幽门螺旋杆菌(HP)的根除治
疗,非常得经典。
以上转载药品安全合作联盟
作者:上海交通大学医学院附属瑞金医院 石浩强
从蛇毒到卡托普利——揭秘“重磅降压药”的发明故事
卡托普利——对于小伙伴们来说,这是一个十分拗口的名字;然而对一些高血压患者来说,它却又是一个耳熟能详的名字!
卡托普利的发明,无疑是高血压治疗史上的一个“里程碑”。你也许会说,听说过蛇毒的药用价值,可从来没听说过降压药的发明还与
蛇毒有关联。
不过,这事还真是真的!从蛇毒到卡托普利的发明,几代科学家为之倾注激情和智慧。这是一场科学探索的“接力赛”,同时又是一场
技术发明的“群英会”!
破译巴西蛇毒的“密码”
说起巴西,也许人们的第一反应可能就是足球,因为巴西是一个享誉全球的“足球王国”。也许还不知道,巴西还有一个令人望而生畏
的景点,那就是“巴西蛇岛”。这个“蛇岛”的面积大约有430平方米,可平均每平方米至少就栖息有9条毒蛇,因此巴西政府严禁人们擅
自进入“蛇岛”。
在这个“蛇岛”上,南美蝮蛇的习性非常神秘,喜欢栖息于灌木丛或者潮湿的雨林中,具有体型大、毒液量多、危险性大等特点。人类
历史上的第一款血管紧张转换酶抑制剂——卡托普利,就是在解析南美蝮蛇的蛇毒“密码”后发明出来的。
卡托普利作为治疗高血压的一项“重磅”级的医药发明,现在已被广泛应用于高血压和某些心脏疾病的治疗。l999年,发明人库什曼和
奥特梯被授予艾伯特雷斯克临床医学研究奖。雷斯克临床医学研究奖是临床医学界的最高奖,是仅次于诺贝尔生理学或医学奖的医学大奖。
看看奖章下有多少“巨人”的肩膀
站在领奖台上的库什曼和奥特梯,无疑是攀登医学科学高峰的胜利者。然而,人类科技发明史上的成功案例大都是一棒接一棒攀登的结
果。那么,在卡托普利成果的幕后,到底还有多少“巨人”的肩膀呢?
其实,医学界早就认识到了蛇毒的降血压作用,并为之进行了卓有成效的探索。1933年,刚毕业的毛里西奥罗查席尔瓦,曾目睹了一
位被巴西蝮蛇咬伤的患者不治而死的全过程。他发现患者发生低血压休克时,老师用了很多升压药都没有把患者的血压升上去。他的老师
也是纳闷,为什么患者的血压升不上去呢?难道蛇毒里含有未知的神秘物质?
老师的疑问让毛里西奥罗查席尔瓦久久不能释怀,难道蛇毒里面存在一种能够降低血压的特殊物质?1948年,他从蛇毒中成功提取了一
个具有肽结构的特殊物质,这种物质就是“缓激肽”。然而缓激肽制剂在血液里的半衰期很短,几分钟就完全失效了,因此没有太大的实
用价值。
1965年,巴西籍博士费雷拉从巴西蝮蛇蛇毒的提取液里找到了一种可以增强缓激肽作用的肽类物质,他把它命名为缓激肽增强因子。
然而,缓激肽增强因子在人体内存在的时间仍然非常短暂。研究到这里,似乎离成功就只有一步之遥了,可是费雷拉却终止了此项研究。
费雷拉转而潜心研究前列腺素在炎症和疼痛方面的作用和机制,后来在该领域取得了一定的成就。
接下来的工作就看罗伯特范恩爵士了。罗伯特范恩可是个医学大家呀,他先后在伯明翰大学、牛津大学学习,曾任英国伦敦皇家外科学
院教授。他由于对前列腺素的研究贡献,获得了1982年的诺贝尔生理学及医学奖。罗伯特范恩以费雷拉带给他的部分蛇毒的干提取物作为
基础,决定进一步开展新型降压药物的研究。
然而,该把这个艰巨的任务交给谁呢?1967年,罗伯特范恩说服了他的同事巴克尔,由他来进行后续的研究。通过用费雷拉的蛇毒提
取液与体外制备的血管紧张素转化酶的反应,巴克尔发现了蛇毒中含有有效的血管紧张素转化酶抑制剂。
尽管蛇毒能够降血压,但是蛇毒并不能直接应用于人体,否则蛇毒会致死人命的。罗伯特范恩还有一个身份,那就是制药公司施贵宝研
究院的顾问。罗伯特范恩深知这项研究的商业价值,因此建议施贵宝公司对蛇毒提取液进行深入研究。尽管施贵宝公司对该研究课题意见
不一,但是有两位实验科学家对该研究课题十分感兴趣。这两位实验科学家就是生物化学库什曼和有机化学家奥特梯。
“登峰”需要勇气和智慧
库什曼和奥特梯都是训练有素的药理学专家,在发明卡托普利的“登峰”过程中表现出了坚韧的毅力和非凡的智慧。
他们制定了周密的科研计划,并一步一个脚印地向前推进。首先从费雷拉的蛇毒提取物中分离出了一种九肽化合物,即含有9个氨基酸
的多肽化合物。这种化合物具有较长的持续作用时间,被命名为替普罗肽。
1971年,由施贵宝出资5万美元合成了1000克替普罗肽。通过在志愿者身上进行的临床试验,证明替普罗肽确实是人体内血管紧张素
转化酶的抑制剂。替普罗肽作为第一代的血管紧张素转化酶抑制剂,不能制成口服制剂进行使用,这是其临床应用的一大障碍。
库什曼和奥特梯决心突破这个障碍,从而为其走向临床铺平道路。他们通过分子修饰研制出了具有较好口服生物利用度的药物,并被命
名为卡托普利。他们通过合成获得的卡托普利口服制剂开始应用于临床,标志着世界上第一个血管紧张素转化酶抑制剂口服制剂诞生了。
1978年以后,商品名为开博通的卡托普利口服制剂开始在市场上销售。
库什曼和奥特梯获得1999年度的雷斯克临床医学研究奖,一是为了表彰他们为发明卡托普利降压药所做出的突出贡献,二是为了表彰
他们的工作开创了一种新的药物开发技术。这种技术就是基于结构的药物设计,即使在现在这种方法仍然十分有效。
没有终点的创新之路
卡托普利降压药在商业上的成功,并没有束缚住科学家进一步创新的脚步。为了获得更好的降压药,默克公司的科学家接过了发明创新
的“接力棒”。
他们通过结构分析和临床试验,终于开发出了依那普利。尽管依那普利是卡托普利的仿制药,但是依那普利具有更好的口服生物利用
度。并且,依那普利还有一个优点,那就是可以延迟药物的作用时间,这样一来一天给一次药就可以达到治疗效果了。
1981年,默克公司完成了依那普利的临床试验,并获得了依那普利的销售资格,商品名为马来酸依那普利。马来酸依那普利作为第二
代血管紧张素转换酶抑制剂,于1985 年 12 月 24 日由 FDA (美国食品药品监督管理局)批准上市。此后,多家制药企业跟进降压药物
的研发,其中普利类的药物也在不断地扩军,从而为心脑血管病的治疗带来了福音。
《中国心血管病报告》指出,2013年中国高血压直接经济负担已增长到2106.5亿元,抗高血压仍然是一项关乎全民健康的战略性课题。
据最新统计数据,2015年中国抗高血压药物市场规模达到252.39亿元,而复方普利类药物的增速居于首位。
参考文献:
【1】品途网.《从敌人到朋友,动物毒素类上市及在研药物盘点》,凤凰网科技,2017-09-14。
【2】陈清奇.《基于模仿创新的小分子药物发明》,科学出版社,2012年版。
转发自: 北京科学技术协会信息中心官方账号
亿珂,伊布替尼成为历史的重磅新药
Imbruvica(亿珂,伊布替尼)最初出现时,有人曾把它称作是“慢性淋巴性白血病(CLL)领域的格列卫”,也有人亲昵地叫它“上
帝的礼物”。
这款改变了血液癌症治疗的重磅药物,背后有着太多不可思议的数字,对于许多患者来说,它能带来接近90%的缓解率;从1期临床到
最终获批,它只用了不到5年的时间;在首次获批的短短14个月内,它总共斩获了FDA的四项适应症批准。2017年,亿珂获得国家食品药
品监督管理总局批准上市。一年后,它又成为了17款被纳入国家基本医疗保险的抗肿瘤药之一。
值得一提的是,这款突破性的新药,与药明康德子公司合全药业有着不解之缘。早在2009年,合全药业就开始生产伊布替尼胶囊的临
床1期用药,并一直支持这款新药从临床试验到获批上市的历程。截止目前,亿珂已在全球90多个国家获批,合全药业也已为其累计生产了
80多吨原料药及中间体。
我们很高兴看到合全药业能够协助这款改变血液癌症治疗的新药来到全球患者身边。在今天的这篇文章中,我们也将介绍这颗杀死血液
肿瘤的子弹,其背后跌宕起伏的研发历程。
1952年,华盛顿沃尔特里德陆军医院的儿科医生Ogden Bruton发现了一种名为“先天性无丙种球蛋白血症”的疾病。通过蛋白质电
泳技术,Bruton医生很快就发现了这些患者都缺乏血清球蛋白。现在,这一原发性免疫缺陷被称为X连锁无丙种球蛋白血症(XLA)。
但直到40年后,XLA的遗传基础才被揭示。在XLA患者体内,B细胞中的一种酪氨酸激酶缺乏表达,导致骨髓中的前体B细胞不能发育
为成熟B细胞并进入外周血,进而导致XLA患者体内所有同种型的血清免疫球蛋白都明显减少或缺失。1993年,两个实验室独立解码了这
种酪氨酸激酶的基因序列,这才将疾病和基因突变联系到了一块儿。
为了纪念发现第一例XLA的Bruton医生,人们以他的姓氏命名了这款酪氨酸激酶(即BTK)。此后,科学家们在更多XLA患者的突变分
析中检测到了超过800种各式各样的BTK基因异常。这些突破性的发现,可谓是了解原发性免疫缺陷遗传基础的里程碑。从那时起,人们开
始关注BTK在B细胞发育和功能中的多种作用。
在随后的几年里,科学家们发现,BTK不仅参与了XLA的发病,还在许多生物学过程中都扮演了重要角色:BTK可以传导,分化和放大
细胞各种表面分子与其微环境通信的信号,同时还可以反过来激活许多主要的下游信号传导途径。
其中,BTK在B细胞受体(BCR)信号传导中的关键作用引起了人们的注意。BCR的信号传导不仅传递与特定抗原接触后的适应性免疫
应答信号,还在B细胞的发育中起到基础作用。通过抗原诱导一系列蛋白质相互作用以及信号分子的募集,BTK会被激活并响应BCR信号,
从而导致B细胞存活,增殖,分化和产生抗体等过程。显然,一旦BTK缺失,BCR信号就不足以诱导B细胞的分化。BTK突变也会造成BCR信
号传导反应异常。
有趣的是,如果B细胞中的BTK过表达,也会引起问题,尤其是炎症和自身免疫疾病。此外,BTK在造血系统的大多数其他细胞中也都
有表达。有研究报道BTK在血小板,巨噬细胞和破骨细胞的发育中也发挥了一定的作用。另一些研究人员指出,BTK参与了一些B细胞进程,
也有可能带来B细胞恶性肿瘤。
由于BTK缺陷主要影响B细胞,BTK也自然成为了自身免疫性疾病和B细胞恶性肿瘤中富有吸引力的一个治疗靶点。很多公司开始针对
BTK的分子结构和功能来设计具有激酶选择性的抑制剂。其中也包括由“科学狂人”J. Craig Venter博士创立的一家名为Celera的公司。
在世纪之交的2000年,Celera公司一度因与美国国立卫生研究院同时完成人类基因组草图而声名大噪。但不久后Venter博士辞职离开,
公司也转而进行新药研发。然而,其大部分尝试都没有成功。随着公司战略目标的再次转移,很多临床前分子也在2006年被转卖给其他公司
,但这段药物研发领域的短暂涉足,却孕育了一款极具潜力,靶向BTK的小分子。
有趣的是,这款小分子最初并非作为药物,而是以“工具化合物”的角色被开发的,它可以与BTK共价结合。由于共价药物可与人体内
蛋白非选择性、不可逆、永久结合,一旦脱靶结合到正常的人体蛋白上,可能引起严重的毒副作用,这种潜在风险使共价药物一度成为药物
研发的禁忌。因此,Celera公司只是把这款分子当作是工具,协助BTK抑制剂的筛选。
虽然这款“工具化合物”在类风湿关节炎等自身免疫疾病中表现出了一定的成药潜力,但Celera最终还是没有坚持开发这款小分子。但
在2006年,一家名为Pharmacyclics的公司,却决定以600万美元,将这款分子以及其他一些候选药物购入囊中。
同年,美国FDA无情地拒绝了Pharmacyclics主打药物的上市申请。一时之间,这家公司陷入了深深的危机。没有人能想到,这款从
Celera斩获的小分子,不仅使公司起死回生,更开启了一段传奇……
从免疫疾病到血液癌症
在Pharmacyclics,这款小分子被命名为PCI-32765(后被称为ibrutinib)。和先前的一些研究所发现的一样,科学家们证实这款BTK
共价抑制剂能够以剂量依赖的方式,显著抑制关节炎的发展。
有意思的是,Pharmacyclics公司当时的首席执行官Richard Miller博士并没有继续关注PCI-32765在自身免疫疾病中的表现,而是意
识到了它在B细胞癌症中的无限潜力。作为一名血液肿瘤的专家,他非常了解,在癌变的B细胞上,BTK蛋白正处于过度活跃的状态。一款
共价抑制剂,或许正是我们所需要的。
在狗的自发性B细胞淋巴瘤中,Richard博士和他的团队首次证实了PCI-32765的体内活性。然而,这种活性表现并不突出,他们所获
得的临床前数据,并不是压倒性的积极结果。这值得花数百万美元来进行临床验证吗?况且,由于这款小分子的共价结合作用,很多研发
人员仍然持否定态度。
促使Richard博士下定决心继续推进研究的,是他的医者仁心。很多患者正在死亡的边缘,亟需更有效的治疗,他不愿意因为对共价机
制风险的不确定担忧,而让这些患者再无止境地等下去。
突破性疗法的诞生
2009年,以ibrutinib之名,PCI-32765的1期临床试验启动了。第二年,Pharmacyclics公司在美国血液学会(ASH)年会上报告了几
项1b/2期试验中,ibrutinib用于各种B细胞恶性肿瘤的概念验证数据。临床初期的积极数据引起了业界的广泛兴趣。不久后,杨森制药也
宣布与Pharmacyclics进行合作,共同研发这款新药。
Ibrutinib也没有令研究人员失望。在一项国际多中心的单臂2期试验PCYC-1104研究中,接受ibrutinib治疗的111名患者疗效显著,总
体缓解率为65.8%,中位缓解持续时间为17.5个月!
基于这项试验的出色数据,ibrutinib获得了FDA的突破性疗法认定。2013年6月末,Pharmacyclics公司向美国FDA递交了新药申请,
11月18日,仅仅四个多月后就获得FDA加速批准上市,单药用于套细胞淋巴瘤!这距离首次进行临床试验也才不到5年。Ibrutinib也有了
更为大家熟知的名字——Imbruvica。
同时,Imbruvica在慢性淋巴性白血病中也表现出了惊人的疗效。2013年12月,在140名慢性淋巴性白血病患者中开展的临床试验显
示,Imbruvica单药治疗后,曾接受过其他治疗和首次接受治疗的患者中,分别有88%和86%出现缓解。这是一个激动人心的结果!对于这
些患者而言,这款全新的药物意味着他们可以脱离对化疗的依赖。
更长久的随访研究表明,Imbruvica单药治疗既往至少接受过一种治疗的慢性淋巴性白血病患者,总体缓解率高达89%,中位无进展生
存期长达52个月,总体生存期达到57%。
随后的两年内,凭借多项临床试验的成功,Imbruvica又快速在FDA获批包括慢性淋巴性白血病和华氏巨球蛋白血症(Waldenstrom
macroglobulinemia)在内的多项适应症。截至目前,已拓展至边缘区淋巴瘤(MZL)和慢性移植物抗宿主病(cGVHD),共有6项适应。
2017年6月,Imbruvica(亿珂)获国家食品药品监督管理总局批准上市,单药用于既往至少接受过一种治疗的慢性淋巴性白血病/小淋
巴细胞淋巴瘤患者以及既往至少接受过一种治疗的套细胞淋巴瘤患者的治疗。距离其在FDA首次获批只间隔了4年。今年10月,亿珂被纳入
国家基本医疗保险目录新药名单。11月,亿珂获国家药监局批准扩大适应症,用于华氏巨球蛋白血症的治疗。随着此次获批,亿珂成为国
内首个也是目前唯一获批用于治疗该疾病的靶向口服药物。
后记
自20世纪末找到导致XLA的BTK基因突变以来,科学家们不但快速确定了BTK在B细胞发育和功能相关的多个重要信号通路中的关键作
用,更是在过去十多年间带来了BTK靶向药物的上市。
随着这些令人兴奋的进展,Imbruvica成为了转化研究的典范。随着这款重磅新药在全球90多个国家的获批,许多B细胞恶性肿瘤患者
的生活迎来了巨大改善。这需要科学家敢于挑战传统的洞见和勇气,也离不开转化医学中来自学术界和产业界的携手努力。我们期待业界能
从成功案例中不断汲取经验,带来更多造福患者的创新药物。
转载:药明康德/报道
意外的惊喜 辉瑞西地那非研发之路
1998年3月,万艾可获FDA批准,成为“勃起功能障碍”的治疗药物,被誉为20世纪90年代男科领域两个革命性进展之一。至今,全
世界120个国家的男性同胞服用了近20亿粒万艾可,万艾可成为了名副其实的“畅销药物”。
万艾可的英文名字Viagra是由Vigor(活力)和Niagra(尼亚加拉)组合而成,尼亚加拉瀑布一直是美洲人甚至欧洲人所熟知的蜜月圣
地,它的“新年头纱”瀑布和雷霆万钧的力量更是闻名遐迩。在中国大陆因注册商标问题还与广州威尔曼公司进行了一场为期十年的官司,
无奈最终被命名为大众并不太熟知的“万艾可”。
万艾可这个“幸福的意外”始于1985年辉瑞计划研发一类可增加心房钠尿肽的降压药物。此时已有研究表明,心房钠尿肽可激发肾中的
环鸟苷酸(cGMP),加快利尿,而磷酸二酯酶5(PDE5)可将cGMP转化为单磷酸鸟苷(GMP),故决定以PDE5作为靶标,开发PDE5
抑制剂降压药。
1989年发现安万特公司的抗过敏药扎普司特舒张肺动脉血管可能与PDE5有关,故以具有轻微PDE5抑制作用的扎普司特为苗头化合物进
行相关研发。随后的三年内辉瑞的5人药化小组经过骨架跃迁和药效团探索,设计并合成了1600个化合物,最终研发出西地那非。
枸橼酸西地那非自1998年上市以来,累积销售额破3000亿,为辉瑞创造了数百亿美元收入,为辉瑞帝国版图的迅速扩张奠定了基础,
也成就辉瑞帝国在全球制药领域的领先地位。
先导化合物的发现
对扎普司特(1)的三唑并嘧啶酮母核进行一系列的变换,发现三唑环变换成甲基吡唑环(2)后,活性和选择性均显著提高,分子模拟
显示,衍生物2与cGMP的的尺寸、形状和偶极距相似度高,故以化合物2为先导化合物,做进一步优化。
先导化合物的优化
分析化合物2母核结构,3位甲基相当于鸟苷酸的戊糖环,履行了核糖片段与受体发生疏水性结合的功能,但体积小,故将其变位正丙基
,得到了衍生物3,活性和选择性进一步提高。5位存在的苯环取代基是必须基团,相当于cGMP磷酸结合位置,1位甲基对PDE5的选择性
很重要,去掉后活性降低(4)。
对5位苯环进行优化发现,利用羟基、硝基、甲磺酰胺基和环丙甲氧基置换乙氧基,活性降低,说明乙氧基是重要的基团。分子模拟也
显示了乙氧基的氧孤电子对与N6的氢原子形成分子内氢键,提高了苯环与母核的共面性,并且连接尺寸大的疏水性基团不利于活性。
化合物3仍为最优结构,但是尚未探索苯环5'位置的取代基对活性的影响,且其脂溶性约为水溶性的10000倍,口服吸收效果差,故在
5'位引入极性基团以提高化合物的成药性。
在苯环上变换或引入基团有三方面的考虑:一是由于这个位置相当于cGMP 的磷酯基,引入模拟磷酯的基团可提高与PDE5 酶的互补性
结合;二是加入极性或成盐基团以有利于水溶性;再一是在研发扎普司特的构效关系时发现,5'位存在有磺酰基或磺酰胺基可提高抗过敏活
性,提示这个位置可容纳新的基团。综上化合物10兼顾了体内活性、选择性和药代性质,故将其制成枸橼酸盐,并命名为西地那非进入临床
研究。
意料之外
枸橼酸西地那非(10)临床Ⅱ试验阶段,心绞痛患者接受治疗后,没能达到预期的效果,因此提前终止了临床试验。
临床试验人员在对西地那非的临床效果进行分析意外发现,一些男性受试者服药后出现生殖器勃起现象,且勃起程度与服用剂量呈正相
关,而高剂量组则有90%的人有勃起现象。
1994年辉瑞以勃起障碍为适应症重启西地那非临床Ⅱ试验,结果显示,12名受试者,有10例受试者勃起功能显著增强。
Ⅲ期临床研究,证实西地那非确实可增强男性勃起的功能,1998年FDA批准其上市。
枸橼酸西地那非的口服生物利用度F=40%,血浆达峰时间为1h,半衰期t1/2 =2h,肝脏中主要经CYP3A4代谢,发生N-去甲基化,去
甲基代谢产物仍具有抑制PDE5作用,但强度只是原药的50%。
西地那非的副作用主要有低血压和蓝视障碍,其中蓝视障碍主要与西地那非抑制PDE6 (IC50=37nmol/L)有关。
小结
尽管辉瑞试图将万艾可粉饰为“缘分和科学”的结晶——“万艾可的发现不是意外、不是偶然,而是创造性思维和研究方向从心绞痛到
勃起功能障碍的重新聚焦”或“对勃起机制更好的理解催生了新型口服药物枸橼酸西地那非”,但这并不能改变它作为“垃圾桶决策模型”
(Garbage Can DecisionProcess)经典案例的命运,即先有解决方案,再去找可以用它来解决的问题并制造需求。
治疗男性勃起功能障碍药物领域,主要有辉瑞西地那非、礼来他达拉非和拜耳伐地那非三大品牌,三大品牌全球销售额合计已超过35亿
美元。目前,他达拉非是世界公认的治疗男性勃起功能障碍最好的药物,其全球销售额自2007年起迅猛增长。2013年全球销售额实现首次反
超西地那非,2017年全球销售额23.23亿美元,远超同年西地那非的12亿美元,2018年受仿制药冲击,这两款原研药销售额均出现下滑,下
降幅度分别为20%和47%,专利悬崖现象凸显。
西地那非是一个原研+仿制的差异化竞争共同做大蛋糕的案例,随着他达拉非的专利到期,外加其长效安全,仿制药上市后势必降低患
者购药成本,带来新的增量。期待越来越多的国内药企加大对他达拉非的布局,挖掘新的机会。
参考资料
1 Terrett NK, Bell AS, Brown D, et al. Sildenafil(Viagra(TM)), a potent and selective inhibitor of type 5 cGMP phosphodiesterase
with utility for the treatment of male erectile dysfunction. Bioorg MedChem Lett, 1996, 6: 1819−1824)
2 Wilson SR, Wilson RB, Shoemaker AL, etal. Antiallergic 8-aza-purines 3. Structural characterization
of2-(2-propoxyphenyl)-8-azahypoxanthine, 2-(2-propoxy-
5-(propylsulfonyl)phenyl)-8-azahypoxanthine,and 2-(2-propoxy-5-(N-methyl-N-isopropylsulfamoyl)phenyl)-8-azahypoxanthine.
J AmChem Soc, 1982, 104: 259−264
3 Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: fromangina to erectile dysfunction to pulmonary hypertension and beyond.
Nat Rev Drug Discov, 2006, 5: 689−702
4 Sung BJ, Hwang KY, Jeon YH, et al. Structure of the catalyticdomain of human phosphodiesterase 5 with bound drug molecules.
Nature, 2003,425: 98−102)
5 郭应禄. 万艾可问世引发的思考[C]. 中国性学会. 中国性学会成立十周年首届中国性科学高级论坛论文汇编. 中国性学会:中国性学会,
2004:13-14.
来源: 药渡 文 | 刘sir
泽布替尼研发历程
2020年百济神州的泽布替尼创造了中国企业原创新药在中美两地同步(时间相隔短于一年)获批的历史。研发客将泽布替尼评选为
2020年度新药,并在本文中回顾其研发历程,希望能为中国产品提供借鉴。
早期阶段:资金法规处处碰壁
泽布替尼是第二代选择性BTK抑制剂,它在百济神州出现属于一个偶然。2012年,现任百济神州全球研究和亚太临床开发负责人的汪
来博士在参加一个会议时,恰巧听到BTK靶点新药的研发进展。会上产品公布的数据相当不错,但汪来发现它并不能对BTK靶点达到完全抑
制,且会与EGFR、HER4、JAK3等靶点结合,疗效和安全性都有改善的空间。
百济神州嗅到了加入BTK抑制剂研发赛道的契机,而且在化合物筛选阶段,即已确定寻找差异化分子的目标——与靶点结合更专一且
抑制率更高。这为之后泽布替尼获得中美两国监管机构认可奠定了基础。
泽布替尼是百济神州成立后合成出来的第3111个化合物,所以产品代号是BGB-3111。但就在化合物刚刚筛选出来的时候,百济神州
出现了资金紧张问题,最困难的时候公司账上仅剩几十万元人民币,连发工资的钱都没有。
然而,百济神州看好泽布替尼的未来,不舍得将这一产品拱手卖掉换钱,所以公司创始人欧雷强决定通过出售公司其他产品的海外权
益来募集资金。最终百济神州在2013年将处于临床前研究阶段的BGB-283(RAF抑制剂)和BGB-290(PARP抑制剂)海外权益相继转让
给默克雪兰诺,将得到的首付款作为泽布替尼研发的资金来源。
此后,泽布替尼开始迈出实现中美同步开发的探索步伐。这条路不用说是对于中国的初创公司,即使对于成熟的跨国公司,在当时中
国的法规环境下也是前途未卜。
早期临床阶段,泽布替尼在国外的研究开展还算顺利,2014年8月在澳大利亚开启首次用于人体的临床研究(FIH),研究代号
BGB-3111-AU-003(NCT02343120),2015年美国也加入这项研究,其他加入的国家还有意大利、韩国和新西兰。但泽布替尼在中国
的临床研究从申请到获批等了一年,2015年2月递交申请,2016年2月才得到临床研究批件,此时国外的研究已于2015年底在美国血液学
会(ASH)年会上公布初步结果。
2020年,研发客年会。研发客出版人戴佳凌给百济神州颁发年度新药奖,汪来博士代表百济神州领奖。更多内容请见第四届研发客创
新药高峰论坛颁出年度9大奖项
注册阶段:天时地利人和
幸运的是,泽布替尼的研发发生在中国药品审评审批制度改革的前夜。
2015年8月国务院发布《关于改革药品医疗器械审评审批制度的意见》(44号文),药品审评资源向创新药倾斜。国际先进的创新药
审评审批理念被国内药品监管机构快速引入,提高审评审批效率,建立创新药加速审评程序、沟通交流机制,接受境外研究数据,一夜之
间这些过去感觉还很陌生的政策词语就在中国迅速落地,成为实现泽布替尼中美同步开发的最关键契机。
百济神州也借机敏锐地将产品开展注册研究的主战场转向国内,利用国外I期研究已经取得的爬坡数据在国内迅速完成桥接试验,紧接
着即在2016年12月启动针对复发或难治性套细胞淋巴瘤(MCL)的关键II期临床研究(BGB-3111-206),此时距离国内I期研究启动才刚
刚过了6个月。而仅一年半后的2018年6月,BGB-3111-206研究就公布研究结果。
原本百济神州就打算以一项I期研究加一项II期研究的方式让泽布替尼尽快上市,恰好遇上国内监管政策改革,加上中国丰富的患者资
源,转向国内开展的II期研究让原来耽误的时间被迅速追赶。
有了天时地利,泽布替尼最终实现中美同步开发的决定性因素则在于其注册团队坚持不懈与中美两国监管部门保持积极沟通。百济神
州决定在中美两国均使用BGB-3111-AU-003和BGB-3111-206的数据作为泽布替尼MCL适应症上市申请的依据。公司与国家药品审评中
心(CDE)的第一次正式沟通交流发生在2018年2月,同年8月百济神州向CDE递交泽布替尼上市申请并获受理,由此推测2月的会议上,
双方已基本达成一致意见。
但说服FDA的过程显得比较艰难。公司注册团队2015年就开始与FDA沟通产品开发计划,直到2018年8月,FDA才明确同意百济神州
提出的做法。不过期间的每次沟通都有收获,在2016~2018的3年间,泽布替尼先后获得FDA授予的“孤儿药认定”、“突破性疗法认定”、
“优先审评”等多 项特殊认定,这些认定也促成了FDA对产品加速获批的认可。
最终泽布替尼的MCL适应症在2019年11月和2020年6月分别获得中美两地上市批准,而且基于相同的研究数据,包括BGB-3111-AU-003
和BGB-3111-206研究共118名MCL患者的疗效和安全性数据,以及BGB-3111-1002、BGB-3111-205、BGB-3111-207、BGB-3111-210
这4个研究共220名各类淋巴瘤患者的安全性数据,除第一个研究以外,其他数据均来自中国患者。
由上可见,泽布替尼实现中美同步开发有几个重要影响因素,首先是在立项阶段选择具有潜在疗效优势的分子作为研发目标,其次恰
好赶上国内药品审评审批制度改革契机,最后是百济神州研发和注册团队嗅觉敏锐,创新产品开发计划敢于向政策借力。这三点成就了泽
布替尼中美同步开发的目标。
如果2015年的药品审评审批制度改革没有发生,泽布替尼要想在中美两地同时上市,很可能需要分别在两国开展注册性临床研究,这
会让刚成立不久的百济神州在资金、人才、时间方面同时承受更大的压力。
WPP子公司KMR集团曾在2016年发布过一项关于临床试验成本的研究,统计了7家大型制药公司在2010~2015年间开展的726项临床
研究成本数据,结果显示各期研究花费的中位值是I期340万美元、II期860万美元、III期2140万美元。
百济神州最终递交的研究数据包括两个I期研究和四个II期研究,若根据KMR给出的花费计算,大约需要4100万美元(约2.7亿人民币)。
如果中美两国分开进行,花费就需要翻倍达到接近1亿美元,而百济神州2016年在纳斯达克上市的募资规模也仅是1亿美元,且据报道公司
上市前手头现金只剩2000多万美元。
百济神州并不像跨国制药巨头那样在全球具有完备的产品研发网络,如果两地分别开展研究实际操作难可能更大,花费也会更大,改
革以前国内各阶段临床研究均要审批才能进行且耗时漫长,以百济神州当时的资金实力很可能无法承担高额花费以及旷日持久的研发耗时。
因此,国内药品审评审批制度改革降低了创新药实现境内外同步开发的成本和难度,除此以外,对于实现产品上市后在国内外的市场
价值也是影响巨大。
改革的意义,可以从竞争产品销售业绩的角度来体会。在泽布替尼上市之前,全球获批上市的BTK抑制剂有强生/艾伯维联合开发的伊
布替尼(Ibrutinib)和阿斯利康的阿卡替尼(Acalabrutinib),两者获批的时间分别是2013年11月和2017年11月。根据公司财报披露,
2019年伊布替尼全球销售额为80.85亿美元,阿卡替尼2018和2019年全球销售额分别为0.642亿美元和1.64亿美元。
如果一开始就按照2020年新版《药品注册管理办法》的注册路径,泽布替尼有可能实现和阿卡替尼几乎同时获批上市。新版《药品注
册管理办法》对于创新药审评,从递交临床研究申请到申请注册上市,都规定了具体的时限。
泽布替尼研发过程中最明显的耽搁是在国内IND申请阶段,此外是上市申请阶段。新版《药品注册管理办法》已将IND审批的时限缩短
到60天,同时明确优先审评的时限为130天,并要求药品检验和生产检查与技术审评同时进行且在审评时限之前完成。这意味着如果泽布替
尼的研究放在现在,中国患者可以直接加入全球多中心I期研究队列,仅这一项就能比原来的研究方案节约1年,同时其在国内的上市申请时
间也可能会大幅缩短。
但审评审批效率提升还是让泽布替尼成为BTK抑制剂的中国第二,上市时间早于阿卡替尼,并在2020年底成功完成医保谈判被列入国
家医保目录。百济神州发布的2020半年报显示,泽布替尼上市后第一个半年的销售收入是769万美元,这一收入完全来自美国市场,预期
产品在中国获批并加入医保目录,2021年可以实现销售额明显提升。
当下国内监管法规对于创新药实现境内外同步开发的障碍已基本消除。根据研发客统计,目前还有近百家国内创新药公司的150多个产
品在国内外同时开展临床研究,其中部分产品亦采用境内外同步开发的办法推进产品研发,如思路迪与康宁杰瑞的KN035、爱科百发的
AK0529。曾经,全球同步开发是高难度的代名词,但在未来将成为常态。
本文转发来自 研发客的雪球专栏 撰文 施樱子
“抗流感神药”达菲(奥司他韦)的前世今生
虽然达菲成名已久,如果不是这场从上月至今肆虐大江南北的流感,很多人可能还不知道达菲/奥司他韦这个药物,可能也不会在
社交网络上对奥司他韦相关的话题产生极大的兴趣。
作为一个医药人,如果进一步深入了解达菲上市前前后后的故事,可能会对制药行业产生全新的认识,当做故事看,非常精彩,堪比
银幕大片,当新药史来研究,你会发现制药行业利润背后的艰辛,无论是研发科学家,还是制药企业、药业大亨,所付出均超出常人所
想。
1.一张海报给科学家了研发灵感
就像苹果砸出牛顿的灵感一样,达菲的研发缘起于一张学术会议海报。
1992年10月14日,在洛杉矶一个学会年会上,吉利德科学公司的一名科学家比朔夫贝格尔(Norbert Bischofberger)博士休息时被一张
海报所吸引。
这张展示药物研发成果的海报内容是一种代号GG167的化合物可以抑制流感病毒在小鼠体内的复制。
这个研究成果由澳大利亚一家生物科技公司 Biota 所拥有,并授权由葛兰素制药开发上市,也就是后来的1999年被FDA批准上市的乐感
清(扎那米韦),也是抗击流感病毒的一种药物,这是后话了。
比朔夫贝格尔看到这个后,就有了自己的不同的想法。
在这之前,抗击流感病毒只能靠两大“金刚”——金刚烷胺和金刚乙胺,不良反应较多,致幻、精神异常什么的,而且容易耐药,GG167
这个药物的确有独特作用,靶点清楚,直接针对流感病毒繁殖的一个酶——神经氨酸酶。
抑制住这个酶的活性,就切断了新病毒和旧细胞的练联系,病毒就没法感染新的细胞,也就抑制了病毒的繁殖。
但是由于GG167的化学结构特点,使得它没法被肠道吸收,所以不能做成口服的,只能做成粉末喷剂,通过吸入肺里起作用,这个不
太方便。
比朔夫贝格尔正是看到了这个缺陷,决心投入到对口服抗流感病毒的药物的研究中。
2. 开发过程一波三折
比朔夫贝格尔请求吉利德公司成立了以自己为组长的研发小组,小组里包括有机化学家、生物学家、计算机专家等,开启了梦想之
路。
现实却不如预期的那般顺利,比朔夫贝格尔的计算机专家多次设计药物分子结构模型,然后有机化学家开始按模型进行化学合成,最
终再做动物实验。
这样的过程多次反复,历经三年,也就是到了1995年底,得到了初步符合要求的化合物GS4071,。
不过令人失望的是,这个化合物经小鼠灌胃给药后,发现并没有被吸收进血液,也就是说,丫和葛兰素的GG167有同样的缺陷!
还好,研发小组没有灰心,继续调整,最终又设计出了新的化合物GS4104,小鼠可以胃肠吸收进入血液,然后再转化成GS4071来产生
抗病毒效应。
3. 动物实验仅仅是第一步
熟悉药物研发的朋友知道,小鼠身上的成功不能宣布药物研发成功,因为还要经过大型动物、人体身上的相继实验才能真正成功。
接着吉利德公司的科研小组相继在大鼠、猴子、雪貂身上进行了实验,结果非常满意!
接下来人体试验如果通过就说明真的有效了,但是到这里遇到困难了,我们知道,人体试验也就是临床试验,非常复杂,像我们现在
的临床试验一般分为I、II、III和IV期临床试验。
耗资高达几个亿美元的临床试验对于当时的吉利德公司是个天文数字,这时必须找合作伙伴了。
4. 罗氏公司投入巨资
1996年1月,吉利德公司开始就转让使用权和多家大药企谈判,罗氏成了最佳选择,同年9月30日,吉利德和罗氏达成协议,将GS4104
的生产和销售权转让给罗氏,罗氏付给吉利德1千万美元继续试验,如果临床试验顺利,罗氏再付4千万美元,正式上市后,罗氏也会分一
部分销售利润给吉利德。
当时,平均5个进行临床试验的药物只有1个能获批,所以这个风险非常大,如果最终没成功,罗氏投入的数千万美元也就打水漂了!
5. 解决生产原料问题
罗氏介入后,发现满足临床试验的原料就是个问题,为啥呢?原来吉利德科研小组使用的以奎宁酸做原料,奎宁酸来自非洲的金鸡纳
树皮,当地兵荒马乱的,来源非常不可靠,当时科研小组的生产工艺方案也就只能100克级的生产水平,大规模临床试验量远远不够!
这时罗氏公司的化学家马丁·卡普夫(Martin Karpf)提出一个新的合成办法,用莽草酸代替,莽草酸这玩意儿来源广泛,很多植物中都
有,尤其是中国盛产的八角茴香里最多,中国进口价格也便宜,所以大批量生产的问题就这么解决啦。
6. 临床试验又遇波折
生产问题解决,此刻万事俱备只欠东风,但是有一个问题也得解决,就是流感和普通感冒的区别,这个不能混淆,混淆了势必影响临
床试验的对象,甚至以后上市后的问题。
于是罗氏召集全球流感专家召开会议,发布流感诊断指南,比如发热、咽喉痛、疲惫乏力等等症状。
这样可以临床试验了,上市前临床试验要做三期,第一期比较简单,效果也不错,这时罗氏按规矩接着付给吉利德4千万美元。
第二期难度略高,需要超过100人的流感病人做受试对象,不过即使100人也难找啊,因为没有流感爆发,这太尴尬啦!!
没办法,只能找了志愿者进行“人造流感”,用浸有流感病毒的棉签插入志愿者鼻孔,如果有感染,接着服药试验,最终结果,效果也
挺满意的,比未用药对象的流感持续时间减少了几乎一半。
下面是最难的第三期临床试验了,为了让药物在上市前更有说服力,此次没有“人造流感”,决定从世界范围内爆发流感的地区招募受
试者。
为了能及时的找到受试者,此次罗氏赞助了世界多个地区的流感预警系统,以便流感爆发时能及时获取开展招募。
可惜老天不配合,世界范围内都没流感爆发,仅在11月25日温哥华地区有流感预警,不过给众多医生配发了药物后,流感并没有爆
发,只有一个患者符合条件,这一切导致整个试验时间延迟,一直拖至1998年6月21日,才基本完成志愿者例数,不过欣慰的是,试验结
果非常理想!
7. 达菲终问世
罗氏1999年3月24日向FDA提交申请,其后把 GS4104 正式命名为达菲,在1999年10月,达菲获得FDA批准上市,从最初到此刻,用了7
年时间,相比较新药平均10~15年研发周期,这时应该可以庆功了。
然而此刻仅仅是上市工作完成了一半,新药上市前五年属于处方药,无法对公众进行广告宣传,而且价格偏贵,有的人认为早两天晚
两天好没大必要,
罗氏的推广团队使用了很多办法,比如在瑞士12个城市组织达菲知识巡回讲座,邀请记者参加新闻发布会,请专家就流感造成的经济
损失进行宣讲等等。
不过难度仍然不小,就像一位罗氏产品经理回忆时说的“七十年来,我们一直告诉人们得流感后最好是卧床休息,而现在,我们要说服
他们去看医生。”
改变人们的认知和就诊习惯一直到现在也是个很困难的事情,不是吗?
还好,当大规模的流感爆发时,人们无须教育,就会立刻认识到达菲这种药物的重大价值,我们现在回顾来看,已经有多次这样的经
过了。
8. 上市后风波不断
不说过去几年,当下不也是有关于达菲/奥司他韦的标题党新闻吗?这样难怪,毕竟达菲/奥司他韦是无可争议的最著名的抗流感病毒
药物。
2003年SARS疫情,达菲在广州卷入一场造谣风波,虽然达菲大卖,但是后来证实,SARS的病原体并非流感病毒也不是禽流感,达菲按
说是无效的,这也导致一些公众和媒体的非议。
到了2005年下半年,禽流感又有全球蔓延趋势,WHO也发警告了,罗氏公司向WHO药品储备库捐赠了300万盒达菲,但是由于全球销
量激增,仍然有很多组织和个人指责罗氏垄断专利。
在这种情况下,虽然专利仍未到期,罗氏公司也宣布放开专利,2005年以后,上药集团和东阳光相继获得生产授权。
大部分药物的上市都有着达菲类似的故事,真的希望公众能够对药企尤其是致力于原研药开发的企业能够给予更多的支持,这样我们
未来才有更好的药物来对付疾病的侵袭,更好的维护公众健康,对于医药人来说,故事背后展现的机遇、创新、勇气、胆识、突破也会给
我们带来很多启示。
本文转载来源: 医药代表 作者: 大咪
第一个重磅炸弹——苯二氮卓类药物的研发史
“重磅炸弹”是属于制药业的一个专属概念,并不是每一个药物都能成为重磅炸弹的。一个药物要成为重磅炸弹,其销售额至少要超过
10亿美元。在制药行业里,能够进入“重磅炸弹”俱乐部的每一个药物,其背后都有一些耐人寻味的故事。上世纪50年代末,由现代药理学
的奠基人之一的瑞典化学家Leo Sternbach博士研发的地西泮,就是历史上第一个年销售额超过10亿美元的“重磅炸弹”药物。
20世纪50年代,随着神经科学和精神病学的快速发展,许多药企开始关注神经精神类药物。1954年Roche作出了一项决定,大力研发镇
静药,并决定要研发出一类药效比现有的镇静剂好且结构新颖的镇静药,Leo Sternbach博士也被分派到这一战略组建的团队中。
由于该领域的研究才刚刚开始,能为他们提供的药理病理方面的信息太少,所以摆在他们面前的只有两条路:要么在已有镇静药的基
础上对其进行结构修饰,要么研发出一类结构全新的镇静药。像安宁、利血平及氯丙嗪就是仿制已有镇静药而上市的镇静剂,要想在此基
础上取得重大突破,实属不易,同时研究者均对药物合成有浓厚的兴趣,所以他们决定走第二条路。
Leo Sternbach博士开始希望从heptoxdiazine 3类化合物开始,因为他在克拉科夫大学做博士后助理期间就是研究该类化合物的,且
该类化合物还具有以下几个特点:①相关的研究较少;②合成难度较小且有一定的挑战性;③结构易于修饰,方便后期的构效关系研究;
④“看”起来具有一定的生物活性。
研究者在heptoxdiazine 3的基础上,对其进行结构修饰,合成了一系列的类似物4,并欲通过引入可能提高生物活性的叔胺基团得到
一系列衍生物5。但是在第一批卤代烷和仲胺合成的40种新化合物中,始终没有发现镇静、抗惊厥的生物活性;而40个药物中的最后一个,
因为团队错拿了甲胺(属于伯胺)代替了仲胺,合成后致使研究团队连将其送去活性筛选的欲望都没有了,命名为Ro-50690后,就直接被束
之高阁,面临被遗弃的命运。
但在对这类叔胺衍生物的进一步研究过程中发现,结构中的氧原子很容易丢失,变成喹啉类衍生物且产率很高,跟heptoxdiazine 3的
母体结构不符,所以研究者进一步猜测,他们合成的这类衍生物的结构式可能是喹啉类氧化物6,该类衍生物结构上的新颖且易得极大的引
起了他们的兴趣,随后他们又合成了一系列该衍生物,但是药理学实验却令人遗憾。但是他们已经有了进一步的设想,即将该氧原子去掉
或还原喹啉环的碳氮双键。
由于几年内,在该领域均未取得重大进展,罗氏上层决定暂时把重心放在抗生素的研发上,所以公司上层解散了Leo Sternbach博士团
队,并要求他们彻底的清扫实验室,他们的进一步设想也就因此而搁浅。在清扫实验台时,研究员Earl Reeder注意到一个命名为Ro-50690
的化合物,并猜想其结构式为衍生物8,大约有一百毫克,其盐酸盐的晶型很好,该化合物早在1955年就已经合成出来了,但是并没有提交
给药理部进行活性研究,于是Leo Sternbach博士抱着试试看的心态,把它送给了药理部,并没有抱多大的希望。但没有多久,药理部反馈
一系列令人振奋的信息:这一化合物有镇静、抗焦虑、松弛肌肉的作用,比市场上的安宁、巴比妥等效果都要好,于是罗氏公司决定以意外
失误而获得的Ro-50690为先导化合物做进一步的深入研究。
由于那时候,核磁和质谱尚未问世,所以他们只能以紫外和红外对衍生物结构进行确定,但是发现化合物8的紫外吸收及红外谱图与原
料7的紫外吸收及红外谱图完全不同,与其他的喹啉类氧化物亦不一样。于是Leo Sternbach博士团队决定对该化合物的元素进行分析,以
便完全确定其结构。
首先他们利用三氯化磷脱掉氧原子后再在酸性条件下,进一步水解得氨基二苯甲酮、甘氨酸及甲胺,而等摩尔量的甘氨酸和甲胺只可能
来自苯二氮卓类衍生物,不可能来自其他结构或同分异构体,所以Leo Sternbach博士确定命名为Ro-50690的化合物应该为衍生物9,而
非衍生物8。反应机理研究显示,可能是氮氧原子的邻位碳原子带正电,更利于甲胺的进攻,而非简单的取代反应。于是Leo Sternbach博
士团队合成了一系列的同类衍生物11,但是活性均比化合物9差。由于衍生物9的活性高且副作用小,临床试验进展非常顺利,并很快生产
上市,命名为氯氮卓又叫利眠宁。临床试验表明,氯氮卓有类似巴比妥的抗惊厥作用,但催眠作用较弱。42家医院中,包括慢性酒精中毒
的众多精神和神经性疾病的患者接受了氯氮卓的治疗。患者的焦虑、紧张情绪得到了显著缓解。氯氮卓可以治疗感情混乱而不会影响到人
的思维能力。帮助人们缓解上瘾情形。1959年,2000名内科医师用它治疗了2万名病人,效果显著。1960年,衍生物9以氯氮卓(Librium)
为商品名在英国上市。随后在世界范围内上市,并且销量节节攀升,迅速成为重磅炸弹品种。
稳定性试验发现,衍生物9的水溶液和混悬液极不稳定,主要原因是2位碳原子及其氨基侧链,所以Leo Sternbach博士决定利用弱酸对
其进行水解,令人兴奋的是水解产物11的镇静作用与化合物9相当,进一步利用三氯化磷去掉氧原子后得衍生物12,其镇静活性略有提高,
这一结构上的改变为后续的构效关系研究拓宽了道路。
在衍生物12的基础上,Leo Sternbach博士及其团队对其进行了全面的构效关系研究,构效关系研究表明:A环:在7位,引入吸电子
基团如NO2、CF3等可增强衍生物的生物活性,而引入给电子基团如甲基、甲氧基等则使活性降低,而在其他的位置引入基团则活性均降
低。B环:在1位上引入甲基则活性增强,而引入更大的基团时则活性降低,如引入正丁基时则没有生物活性。C环:在2′位引入卤素时(如
F、Cl)则使衍生物的活性增强,而在4′引入取代基则活性降低。
罗氏公司在研发苯二氮卓类镇静药的过程中,合成了3000个相关的化合物,但只有氯氮卓9和地西泮13两个获得了成功。1963年,
Diazepam (Valium安定)地西泮上市,成为了历史上第一个年销售额10亿美元的“重磅炸弹”药物。谈起这一首个重磅药物,美国报纸说
它使得充满焦虑的美国主流社会以半娱乐心态服用药物的文化获得松绑。
惠氏公司也在氯氮卓的基础上,开发出了奥沙西泮(Oxazepam)。而睡眠障碍由硝基安定治疗,商品名Mogadon,于1965年引入。替
马西泮于1969年引入。盐酸氟胺安定,商品名Dalmane,于1973年上市。到了1983年,共有17种苯氮卓类药物上市,年销量超过了30亿,
而今天已经有29种同类药物上市了。对于焦虑、失落、失眠和压力大等症状,药物需要长期服用,甚至要数年。到1970年代,苯氮卓类药
物成为临床上最常用的处方药,估计有5分之一的女性和10分之一的男性都服用它们。
总之,地西泮(Diazepam)正是研究者正视偶然化学实验失败和偶然的失误而执着开发成功的镇静药,是历史上第一个年销售额超过10
亿美元的“巨磅炸弹”级的药物,同时也是抗焦虑活性的“金标准”。历史总是充满着偶然与必然,但机会是留给那些有准备的人;而好
奇心、观察力、创造力与探索精神成就了传奇药物,成就了传奇的科学人生。也正因为地西泮及不断涌现同一家族药物使Roche公司进入
制药工业巨人的行列。正所谓千淘万漉虽辛苦,吹尽狂沙始“安定”。
资料来源:
Leo H. Sternbach. The Benzodiazepine Story. Journal of Medkina1 Chemistry,1978,22 (1): 1-7.
本文来源: 药渡 作者:人成言
华法林的发现史:从灭鼠药到救命药
在过去的近半个世纪,华法林一直是口服抗凝药物的代表和支柱,广泛用于血栓栓塞性疾病、心房纤颤、人工瓣膜置换术后等领域,是
口服抗凝药物中的“霸主”。据统计,每100个英国人,至少有1人长期使用华法林,而在老年患者中比例更高。但随着达比加群酯、利伐
沙班等新型口服抗凝药物的出现,可以为患者提供更安全和方便的治疗,华法林的“霸主”地位逐渐受到挑战。在华法林由盛转衰的重要
转折时刻,作为见证者,我们有必要对这一划时代药物的发现历史进行回顾,在致敬华法林发现者的同时,深思其带给我们关于科学研究
规律的启示。
1 甜苜蓿病
20世纪20年代,北美洲大草原的牧民发现了一种奇特现象:牲畜进食发霉的甜苜蓿干草后约15 d会有出血表现(轻微外伤后出血不止,
甚至会发生自发性出血,血液难以凝固),并在30~50 d内死亡。1924年,加拿大兽医 Frank W.Schofield在《美国兽医学学报》(Journal
of the American Veterinary Medicine Association)详细描述了这种疾病,并认为该病可能与发霉干草中的黑青霉等有关。由于与甜苜蓿
干草有关,该病也被称为“甜苜蓿病”。
经过近10年的研究,人们发现:避免牲畜食用发霉的干草,或对发病后牲畜进行输血治疗,对甜苜蓿病有一定防治效果,但由于病因
尚未破解,在整个20世纪30年代,该病对北美畜牧业仍构成很大威胁。这一时期,席卷世界的经济危机正给美国人民的生活带来极大困扰,
病因尚未破解的“甜苜蓿病”疫情导致大量牲畜死亡和经济损失,这对北美牧区的居民无疑是雪上加霜。
在损失了牲畜的居民中,有一位名叫Ed Carlson的农夫,在一场暴风雪中,他用卡车载着一头死亡的奶牛、一桶来自于死亡奶牛的不凝
血、约45 kg甜苜蓿干草,跋涉约300 公里,来到了当地一所不起眼的农业试验站,在那里他偶遇了1个月前刚刚开始研究“甜苜蓿病”的
生化学家Karl Link,从而开启了发现华法林的波澜壮阔之旅。
2 Karl Link教授生平
Karl Link教授(1901—1978年)生于美国印第安纳州的拉波特市,父亲是一位路德教牧师。Karl Link于1918至1925年就读于威斯康
辛大学农业学院,并于1925年获得了农业化学博士学位。随后,他在美国国家教育委员会的资助下,前往欧洲进行博士后培训,曾在苏格
兰和瑞士进行研究并先后与Fritz Pregl(1923年诺贝尔化学奖得主)和Paul Karrer(1937年诺贝尔化学奖得主)共事。在完成欧洲的培
训后,Karl Link于1927年回到美国威斯康辛大学谋得教职,遗憾的是,Karl Link在瑞士期间罹患肺结核,因当时结核病治疗手段相对落后
,他一直受疾病困扰,直至去世。
在那个风雪之夜,Ed Carlson送来的死亡奶牛、不凝血和发霉的干草,为Karl link教授提供了珍贵的研究材料。经过6年的不懈努力,
1941年,他终于发现了“甜苜蓿病”的元凶:甜苜蓿中含有的天然香豆素,虽然香豆素本身并不具有抗凝作用,但在真菌(发霉)的作用
下会被氧化为双香豆素,干扰维生素K依赖性凝血因子的功能,引起出血。经济向好时,牧民们一般不会将发霉的草料喂给动物,而是重新
更换干燥的草料,因此“甜苜蓿病”的问题并不突出;但在经济萧条时期,牧民们无力替换发霉饲料,该问题即突显出来。至此,困扰北美
牧区近20年的怪病之谜得以完美解答。
3 从灭鼠药到口服抗凝药
第二次世界大战期间,美国鼠患严重,亟需有效的灭鼠手段。1945年,因“胸膜炎”住院休养的Karl Link想到了采用双香豆素的衍生
物作为灭鼠药的主意。因双香豆素起效慢,患“甜苜蓿病”的牲畜30~50 d后才死亡,所以Karl Link与其合作者在筛选了上百种双香豆素衍
生物后,最终在1948年发现了苄丙酮香豆素并将其作为理想的灭鼠药。由于这项艰苦的研究工作是在威斯康辛大学校友基金会(WARF)
的资助下完成的,所以将这种香豆素衍生物命名为Warfarin(WARF:基金会;-arin:香豆素词尾),中文译为华法林,沿用至今。作为
灭鼠药,华法林取得了巨大成功,也是后续效果更强的灭鼠药溴敌隆(又称“超级华法林”)的原型药物。
随着华法林在灭鼠事业上的巨大成功,逐渐有医师开始尝试在临床中使用该药用于抗凝。因曾用作“灭鼠药”,患者在接受该药上存在
一定困难。转机出现在1955年,二战英雄、五星上将、时任美国总统的艾森豪威尔在打高尔夫球后出现心肌梗死,随后接受了华法林治疗。
这一事件增加了民众对华法林的接受程度,于是逐渐发生了我们所熟知的华法林用于临床治疗的诸多故事。尤其是在20世纪80年代,世界
卫生组织推荐采用国际标准化比值监测华法林疗效后,克服了制约华法林广泛应用的剂量控制问题。自此,在全世界范围内,华法林正式成
为使用最广泛的口服抗凝药物。
4 历史的启示
回顾华法林的发现史,我们饶有兴趣地发现许多貌似偶然的事件堆叠:经济寒潮令牧民无力更换发霉的牧草;绝望的农夫突发奇想,冒
雪驱车数百公里,在不起眼的地方农业试验站偶遇一位刚刚于1个月前才开始对“甜苜蓿病”感兴趣的生化学家;由于罹患疾病,不得不住
院休养的生化学家研究起了灭鼠药物;全民英雄、美国总统在任期内突发心肌梗死并从该药获益。
然而,偶然中总是存在着必然,伟大的创新背后是厚积薄发:美国从1875年开始建立起遍布全美的农业试验站体系,重视基础科研和
农业应用,为Ed Carlson与Karl Link历史性会面创造了机遇;虽然威斯康辛州不是美国经济发达地区,却因重视教育,伫立着一所始建于
1848年、世界综合大学排名前100强的著名大学威斯康辛大学,这所大学不仅为Karl Link教授提供科研场所,其校友基金会也恰好为华法
林的研究提供了经费支持,并为写入医学史的该药物冠名;虽然Karl Link罹患肺结核,但他坚持不懈、不断尝试,即便在住院休养期间也
心系研究。华法林的故事,不但为我们揭示了科学研究从现象(牲畜出血死亡)到本质(双香豆素抗凝作用)、从实验室(发现双香豆素
的化学结构,并合成其衍生物)到实践(灭鼠药)及临床应用(口服抗凝药“霸主”华法林)的一般规律,还提示我们,良好的基础教育
和基础科研条件,以及充分的经费支持,是催生划时代意义发现的重要条件。
5 结语
作为目前最重要的口服抗凝药物,随着新型口服抗凝药物的不断涌现,华法林时代或许已经开始由盛转衰。当我们怀着对先辈深深的敬
意,在这个特殊的时间节点回顾华法林的发现史,为我们未来的科学研究带去有益启示。
原文作者:
张路 北京协和医院血液内科
首发于“xieheyixue”协和医学杂志微信
X射线的发展历程
X射线的发现是19世纪末20世纪初物理学的三大发现(X射线1896年、放射线1896年、电子1897年)之一,这一发现标志着现代物理学
的产生。X射线的发现为诸多科学领域提供了一种行之有效的研究手段。X射线的发现和研究,对20世纪以来的物理学以至整个科学技术的发
展产生了巨大而深远的影响。
失之交臂
1836年,英国科学家迈克尔.法拉第(Michael Faraday,1791-1867)发现,在稀薄气体中放电时会产生一种绚丽的辉光。后来,物理
学家把这种辉光称为“阴极射线”,因为它是由阴极发出的。
1861年,英国科学家威廉.克鲁克斯(William Crookes,1832-1919)发现通电的阴极射线管在放电时会产生亮光,于是就把它拍下来,
可是显影后发现整张干版上什么也没照上,一片模糊。他以为干版旧了,又用新干版连续照了三次,依然如此。克鲁克斯的实验室非常简陋,
他认为是干版有毛病,退给了厂家。他也曾发现抽屉里保存在暗盒里的胶卷莫名其妙地感光报废了,他找到胶片厂商,指斥其产品低劣。一个
伟大的发现与他失之交臂,直到伦琴发现了X光,克鲁克斯才恍然大悟。
在伦琴发现X光的五年前,美国科学家古德斯柏德在实验室里偶然洗出了一张X射线的透视底片。但他归因于照片的冲洗药水或冲洗技
术,便把这一“偶然”弃之于垃圾堆中。
发现X射线
1895年10月,德国实验物理学家伦琴(Wilhelm Konrad Rontgen,1854~1923)也发现了干板底片“跑光”现象,他决心查个水
落石出。伦琴吃住在实验室,一连做了7个星期的秘密实验。11月8日,伦琴用克鲁克斯阴极射线管做实验,他用黑纸把管严密地包起来,
只留下一条窄缝。他发现电流通过时,两米开外一个涂了亚铂氰化钡的小屏发出明亮的荧光。如果用厚书、2-3厘米厚的木板或几厘米厚的
硬橡胶插在放电管和荧光屏之间,仍能看到荧光。他又用盛有水、二硫化碳或其他液体进行实验,实验结果表明它们也是“透明的”,铜、
银、金、铂、铝等金属也能让这种射线透过,只要它们不太厚。使伦琴更为惊讶的是,当他把手放在纸屏前时,纸屏上留下了手骨的阴影。
伦琴意识到这可能是某种特殊的从来没有观察到的射线,它具有特别强的穿透力。伦琴用这种射线拍摄了他夫人的手的照片,显示出手的骨
骼结构。
1895年12月28日,伦琴向德国维尔兹堡物理和医学学会递交了第一篇研究通讯《一种新射线——初步报告》。伦琴在他的通讯中把这
一新射线称为X射线(数学上经常使用的未知数符号X),因为他当时无法确定这一新射线的本质。
伦琴的这一发现立即引起了强烈的反响:1896年1月4日柏林物理学会成立50周年纪念展览会上展出X射线照片。1月5日维也纳《新闻
报》抢先作了报道;1月6日伦敦《每日纪事》向全世界发布消息,宣告发现X射线。这些宣传,轰动了当时国际学术界,伦琴的论文在3个
月之内就印刷了5次,立即被译成英、法、意、俄等国文字。X射线作为世纪之交的三大发现之一,引起了学术界极大的研究热情。此后,
伦琴发表了《论一种新型的射线》、《关于X射线的进一步观察》等一系列研究论文。1901年诺贝尔奖第一次颁发,伦琴就由于发现X射线
而获得了这一年的物理学奖。
伦琴发现X射线使X射线研究迅速升温,几乎所有的欧洲实验室都立即用X射线管来进行试验和拍照。几个星期之后,X射线已开始被医
学家利用。医生应用X射线准确地显示了人体的骨骼,这是物理学的新发现在医学中最迅速的应用。随后,创立了用X射线检查食道、肠道
和胃的方法,受检查者吞服一种造影剂(如硫酸钡),再经X射线照射,便可显示出病变部位的情景。以后又发明了用于检查人体内脏其他
一些部位的造影剂。X射线诊断仪在相当一个时期内一直作为医院中最重要的诊断仪器。
为纪念伦琴对物理学的贡献,后人也称X射线为伦琴射线,并以伦琴的名字作为X射线等的照射量单位。
本文转载摘自中国科学院高能物理研究所
麻醉剂的发明和出现
19世纪40年代在使用麻醉剂之前,惨死在外科医生刀下的人所经受的无可名状的痛苦无疑是存在的。但是,我们绝不该认为,在现代外
科学问世之前,每一例手术都伴随着令人感到毛骨惊然的痛苦嘶叫。人类的一个早期愿望必定是对魔幻止痛物的希冀。为达到这一目的,古
代的医生们对某些植物的止痛性能作过广泛的研究,从实践经验中悉心积累了大量的知识。
古代西方麻醉剂起源
至少在4000年之前,可卡因(在南美洲,从古柯植物中提取)和鸦片(在近东地区,从罂粟花中提取)就已作为影响心理状态的药品而为人所
熟知。它们似乎也在同样早的年代里被当作药物来使用。罗马的医生们掌握着若干种可当作止痛药和安眠药来使用的毒品。鸦片是使用最为
普遍的药品之一:公元1世纪初,作家塞尔苏斯曾建议通过饮用野生罂粟花的汁液来治疗"头痛、溃疡、眼炎、牙疼、呼吸困难、肠道疼痛、
子宫痉挛以及滚关节、肝区、脾脏和肋骨疼痛"。他还谈到了将莨菪当作镇静剂使用的情况。在德意志西部诺伊斯一地的古罗马军医院中,
曾发现大量的莨菪种子,以及其他草药。莨菪含有东莨菪碱。这种药物至今仍被当作术前麻醉剂来使用--少量服用可诱发睡眠和记忆缺失。
对罗马人来说,药力最强的麻醉剂当属曼德拉草。这是一种名声可怕的植物。尽管很难确定古代文字记载中有关曼德拉草的所有描述指
的是否都是同一种植物,但将药用曼德拉草确认为曼陀罗子,其理由还是充分的。曼陀罗子含有颠茄碱和东莨菪碱,都是减缓心律的药物,
服用得当还能彻底消除疼痛,减少手术给病人带来的精神创伤。在阿兹台克人那里,曼陀罗子茶是一种供献祭者食用、使之进人昏睡状态,
以便忘却自己最终结局的物质。普林尼于公元75年前后描述过罗马医生更具建设性地使用这种药物的过程:"在把曼德拉草当作安眠药水来
使用时,规定的剂量应当同患者的强壮程度构成比例,中等剂量为一塞亚图斯(约为三汤匙)。用水冲服可医治蛇咬,在做外科和穿孔手术之
前服用,还可起到麻醉作用。"
到普林尼在世时,在经过艰苦探索之后,过量服用这种药物所带来的危险显然已为人们所了解。或许是由于有种种危害,因此,只有在
绝对必要的情况下,而非例行诊治时,才能使用这类药物。大部分小手术都不用麻醉剂。塞尔苏斯就此指出,优秀外科医生所具备的一项
条件是专注于工作而不为患者的叫喊所动摇的能力。
西方中世纪麻醉剂发展
在欧洲,为古罗马人所熟知的这些麻醉药物直到中世纪还在继续使用。但在摄取途径方面发生了很大的变化。最常见的摄取途径是公元
9-15世纪无数典籍中提到的"催眠海绵"方式。这些药物--包括鸦片、曼德拉草、莨菪和从芹叶钩吻中提炼的毒物--经混合后被浸入海绵之中
,随后将海绵晾干。在需要麻醉剂时可将海绵浸湿,然后放在患者的嘴上,让患者吸入药味。这些混合药物肯定会使任何人陷人毫无知觉
的状态,尽管吸人致命的芹叶钩吻毒物(可以先后抑制神经系统的运动中枢和感觉中枢),会使整个手术极具风险。
在为口腔麻醉开出各种处方时,类似的问题也会出现。15世纪的很多手抄本对此都有描述。事实上,每种处方通常都只有几个副本(往往
只有一个)。这种情况表明,这些处方并不像所说的那样深受欢迎--估计是因为它们在剂量过高时可能会带来致命的危险。但是,英格兰人
为一副称作"德韦尔"的饮剂开出的处方却好像给出了正确的配方。已知的将近30个副本都给出了完全相同的成分和配制方案--混合到一起,
煮开一小段时间,然后倒入酒中稀释。除了其他麻醉处方所包括的芹叶钩吻汁液、鸦片和莨菪外,这一处方还含有醋剂和泻根。前者可以
抵消芹叶钩吻的毒副作用,后者则与莨菪同为强力泻剂。在喝下这副饮剂之后,患者好像会迅速失去知觉,给施行截肢、排脓或缝合伤口
等简单手术留下足够的时间。随后,泻根和芹叶钩吻的轻泻作用,会使患者在一段时间内产生很不舒服的感觉,但肯定能将其他成分驱除
出去。这些成分如果继续滞留在系统之内,则肯定会带来致命的后果。从它深受欢迎的情况判断,"德韦尔"(在中古英语中,它的含义类似
于"无知觉")饮剂肯定是相当可靠的。那些手抄本对它的用途作了奇特而有趣的描述:"在需要它的时候,让准备开刀的人背靠大火坐下,并
动员他喝下(德韦尔饮剂),直到入睡,然后你可以放心地给他开刀,等治疗完毕并想让他醒来时,拿些醋和盐擦洗他的太阳穴和颧骨,他就
会立刻苏醒过来。"
古代中国麻醉剂
中国古人很久以前就有关于手术麻醉的传说和记载,例如"神农尝百草,一日而遇七十毒",就反映了中国古代人民很久以来就千方百计
寻找治病止疼的良药。公元2世纪,我国伟大的医学家华佗发明了"麻沸散",1700多年前,华佗就已经使用全身麻醉进行腹腔手术。而欧
美使用全身麻醉术是十九世纪初的事,比我国推迟了一千六百多年。
华佗是我国医学史上第一个施行剖腹手术的外科医生。有一次,华佗看见一个面色焦黄、呼吸紧促、病势很重的推车人,一问,才知他
肚内绞痛。华佗断定这人得的是肠痈(即阑尾炎)。于是华佗让病人喝了麻沸散,很快就麻醉过去。他用刀子切开腹部,割去肠子溃烂部分,
洗涤内部,然后缝和起来,又涂上消炎生肌的药膏。过了几天,病人的伤口很快就愈合了。这一手术的成功,不仅说明了麻沸散的效力,
而且也说明了华佗在人体解剖上有丰富的知识。
公元652年,孙思邈著《备急千金药方》,1596年李时珍在《本草纲目》中,介绍了曼陀罗花的麻醉作用。1743年赵学敏所著《串雅内
编》介绍了由草乌、川乌、天南星、蟾酥、番木鳖等组成的开刀药方。在复苏急救方面,公元前4-5世纪,就有扁鹊切脉以诊断人之生死,
用针和草药进行急救复苏的记载。
本文转载自 百拇医药网
毒树、凶器、天然抗癌药典范——紫杉醇的前世今生
来源 | DrWhy
作者 | WXY707
1943年12月2日晚,意大利东南部的巴里港灯火通明,工人们被要求全力加速卸载货船上的军用物资,因为盟军打算尽快备齐物资以
便挺进意大利北部并攻占罗马。然而,令盟军没有想到的是,德军在当晚发动了空袭!
松懈的防卫加上港口的照明,让德军轻轻松松炸毁了停泊在港口的多艘船只。更糟糕的是,被击毁的一艘名为“约翰·哈维”的美国商
船上,秘密装载了2000枚芥子气炸弹!
原本美军为的是应对德军可能的毒气战,结果这次空袭却造成了有史以来最严重的毒气泄漏事件,让许许多多的军人和平民无辜伤亡。
不久之后,负责调查此事的美军化学战顾问亚历山大发现,芥子气破坏了人体的大部分白细胞。
这给耶鲁大学的古德曼(Louis Goodman)和吉尔曼(Alfred Gilman)两位药理学家带来了重大的启示,他们据此开发出来了第一个
肿瘤化疗药物——氮芥[1]。
二战之后,美国加快了抗癌药物的筛选工作。1955年,美国国家癌症研究中心(NCI)成立了癌症化疗国家服务中心(CCNSC),专
门负责全国性的抗癌药物筛查工作[2]。
一开始,这个机构主要是评估化学结构已知的化合物。到了1960年,他们开始从动植物的天然提取物中筛选抗癌药物。为此,NCI还专
门跟美国农业部达成了合作协议,由农业部负责向NCI提供植物样本用于抗癌药物的筛选。
此后的20年间,研究人员总共收集并测试了3万多个样本,就是在这样的规模浩大的抗癌药物筛选工程中,一个迄今为止最优秀的天然
抗癌药物诞生了!
它就是紫杉醇!
一棵不起眼的太平洋紫杉
1962年8月份的某一天,美国农业部植物学家亚瑟·巴克雷(Arthur Barclay),带着他的3个研究生,顶着大太阳,来到华盛顿州的吉
福德·平肖国家森林收集植物样本。无意间,巴克雷在一小片针叶树中发现了一棵7米多高的太平洋紫杉。
看着这棵不起眼的紫杉,巴克雷没多想就把它命名为B-1645,因为这是他采集到的第1645个植物样本。接着,巴克雷让他的研究生收
集了这棵紫杉的树枝、树皮和果实样本[3]。然后,师徒四人乐呵呵回到农业部交差去了。
按照农业部的要求,他们把采集来的样本寄给了NCI。之后,他们并没有把这件事放在心上,该干嘛干嘛去了。只是,他们不知道,这
棵不起眼的太平洋紫杉就要改变历史了!
太平洋紫杉,又叫短叶红豆杉,是红豆杉科红豆杉属的植物。红豆杉距今已经有250万年的历史了,因此又被称作是植物王国的“活化
石”。
其实,人类在很早之前就开始利用红豆杉了。不过,它不是被用作救人的药物,而是被用作杀人的武器!曾经在中世纪欧洲征战中大出
风头的英国长弓,就是用紫杉木制成的!
而这一次,紫杉却注定要一改从前杀人如麻的狰狞面目,转而成为救人水火的抗癌良药!
NCI在接到太平洋紫杉样本之后,就开展了相关的研究工作,初步的研究结果显示,树皮中的粗提取物对人口腔表皮样癌(KB)细胞有
细胞毒性作用[4],这似乎意味着紫杉树皮中含有某种抗癌物质。
于是,NCI要求农业部再多提供一些太平洋紫杉树皮。这个光荣的任务自然落到了巴克雷头上。1964年9月,巴克雷回到吉福德·平肖国
家森林,索性一下子收集了二十多斤树皮[3],寄给了NCI。
这些宝贵的原材料辗转又到了北卡三角研究院,在这里,它与两个著名的化学家相遇了,他们就是沃尔(Monroe Wall)和瓦尼(Mansukh Wani)。
紫杉醇的诞生
沃尔和瓦尼在收到这些树皮后,就展开了分离提纯工作。他们小心翼翼地把这些树皮研磨、一步步地进行提纯,并用一种白血病小鼠模
型验证各个步骤中提纯物质的抗癌活性。
1966年9月,沃尔和瓦尼经过反复地尝试后,终于提纯得到了一种编号为K172的物质,它具有很好的抗癌活性。因为当时还没有搞清
楚这种物质的具体结构,但他们知道这种物质肯定含有羟基(醇类所含有的基团);又因为它是从太平洋紫杉(Taxus brevifolia)中获得
的,所以,沃尔把这种物质命名为紫杉醇(taxol)[4]。
不过,沃尔和瓦尼从12公斤紫杉树皮中只提纯得到了0.5克的紫杉醇,收率非常低,只有0.004%[4]。这就给紫杉醇后来的曲折研发历
程埋下了伏笔。
1967年,在美国化学会的年会上,瓦尼做了报告,跟大家展示了紫杉醇的细胞毒性和抗肿瘤活性研究结果。
但是,由于紫杉醇的结构还没有搞清楚,并且它的提纯产率非常低,又因为NCI在那个年代筛选了一大批抗癌药物,这其中就包括沃尔
和瓦尼发现的喜树碱,而且NCI在那时已经在准备开展喜树碱的临床试验了。
所以,沃尔打算暂时把紫杉醇放一放。不过,瓦尼不同意,他坚持在“低优先级”的状态下,继续开展紫杉醇研究[4]。
瓦尼的坚持给紫杉醇带来了活路,终于在1969年,紫杉醇通过了NCI严格的抗癌药物筛选程序,成为了官方认可的潜在抗癌药物。
1971年,沃尔和瓦尼等人在材料与化学领域的顶级期刊《美国化学会杂志》上发表了一项重磅研究,他们采用X射线衍射和核磁共振分
析,确定了紫杉醇的结构[5]。
原来紫杉醇是由47个碳原子组成的环状化合物,总共有11个立体中心,还有很多个功能团。总之,结构极其复杂,简直就是大自然调
制出的“小怪物”!
在这篇论文中,沃尔和瓦尼还指出,这种复杂的天然化合物,在初步的试验中显示出抗白血病和抗肿瘤的效果。
然而,这篇文章的发表,并没有让紫杉醇迅速进入临床试验。相反,它却引起了环保主义者的注意。
抗癌与环保之争
在沃尔和瓦尼公布紫杉醇的分子结构之后,NCI的管理层似乎对紫杉醇有点感兴趣了。不过,紫杉醇的供应却是他们首先考虑的问题,
因为要开展紫杉醇的动物试验、毒理实验和以后的临床试验,都需要大量的紫杉醇[4]。
所以,沃尔和瓦尼接下来的主要工作就是从紫杉树皮中提取紫杉醇,而美国农业部也不得不向他们提供大量的紫杉树皮,这就意味着
有许许多多紫杉树要被砍掉。
原本太平洋紫杉是不起眼的植物,而美国农业部却突然间大量砍伐紫杉,这让环保人士心里有些犯嘀咕:他们这是为什么?
与此同时,紫杉醇的抗癌机制研究也在不温不火地进行着。1978年,NCI的两名研究人员发现,紫杉醇能够让细胞停滞在G2-M期,抑
制细胞的有丝分裂[6]。这似乎意味着,紫杉醇跟秋水仙碱、长春花碱、美登木素等一样,都是一类具有微管蛋白抑制作用的药物。
然而,直到1979年,分子药理学家苏珊·霍尔维茨(Susan Horwitz)的研究[7],才真正揭开了紫杉醇抗癌的“秘密花园”!
霍尔维茨发现,紫杉醇可以稳定和增强微管蛋白的聚合,而这刚好跟上面提到的药物作用相反,因为它们都是抑制微管蛋白的聚合。
紫杉醇促进微管蛋白聚合、防止微管的解聚,这就会让细胞在分裂过程中被卡住了,所以细胞就会发生凋亡,紫杉醇的抗癌作用也就得
到发挥了。
在霍尔维茨宣布了紫杉醇抗癌机理之后,NCI终于下定决心把紫杉醇列为重点开发对象了。
1984年,紫杉醇的I期临床试验开展。次年,II期临床也开始启动了。1988年,NCI首次公布II期临床结果[8],发现紫杉醇对黑色素瘤
的疗效非常显著,更重要的是,它对复发性卵巢癌的有效率达到了30%!
这在当时简直是爆炸性新闻,因为那个时候,对于复发的卵巢癌还没有什么有效药物,紫杉醇一跃成为明星药物,太平洋紫杉也一下子
从遭人嫌弃的有毒树木变成了惹人关注的抗癌药材。
而环保主义者们,这个时候就更加关注NCI的动态了。按照NCI的一名官员推算,要想用紫杉醇治疗全美国的黑色素瘤和卵巢癌患者,
那么每年需要砍伐36万棵紫杉树才行[3]。
这番言论让环保主义者们更加坐不住了。他们开始抗议,认为太平洋紫杉生长缓慢,天然更新的能力很差,如果大量砍伐,紫杉是要灭
绝的。而且,他们还认为,长有紫杉的森林是一种濒临灭绝的猫头鹰栖息地,如果肆无忌惮地砍伐紫杉树,失去的将不止是一个物种!
所以,环保主义者要保护树木,而肿瘤专家和患者需要药物,救树还是救人就成为了当时社会各界热议的话题,经过媒体的报道,紫杉
醇和太平洋紫杉一度成为了政治事务。
这种焦灼的状态,随着紫杉醇的合成才最终得以解决。
紫杉醇的合成
在20世纪七八十年代,紫杉醇结构上的复杂性吸引了全世界化学家的目光。一时间,从头合成出紫杉醇成了许多化学家们证明自己实
力的一种选择。
90年代初期,已经有很多研究小组[9, 10]采用不同的路径合成了紫杉醇,但这些方法动辄要几十步、过程复杂且反应条件苛刻,而且
合成所需的试剂非常昂贵,合成的总体收率也非常低。所以,紫杉醇的全合成虽然能代表合成技术的成就,但并不适合于工业化生产。
真正让紫杉醇化学合成进入工业化生产的方法是半合成途径。
1981年,法国科学家Pierre Potier,从英国紫杉的叶子中分离得到了一种叫做10-DAB的物质,它跟紫杉醇的结构非常相似,只是缺少
一些侧链基团。而且,10-DAB在英国紫杉叶片中的含量比较高,叶子还可以再生,不会对紫杉树造成大的影响[11]。
那么,能不能用10-DAB作为底物合成紫杉醇呢?
带着这样的疑问,Potier带着他的团队经过多年的努力,终于在1988年的《美国化学会杂志》上公布了采用10-DAB合成紫杉醇的方法
[12],这种方法的总产率超过了50%!
论文的发表立即引起了不少化学家的注意。于是,也有不少化学家改进、优化了半合成的方法,这给紫杉醇的工业化生产提供了可行的
方法。
1991年,百时美施贵宝(BMS)与NCI达成合作协议,由BMS负责紫杉醇的商业化生产,BMS采用的就是从10-DAB合成紫杉醇的半
合成方法[4]。这种方法也让环保主义者们不再担心太平洋紫杉的问题了。
终于,在1992年12月29日,FDA批准紫杉醇注射液,商品名为泰素(Taxol),用于治疗晚期卵巢癌[3]。之后,紫杉醇又被批准用于
治疗乳腺癌、非小细胞肺癌、卡波西肉瘤等癌症。
如今,紫杉醇已经是鼎鼎有名的化疗药物。然而,这不是它的终点,由紫杉醇研究衍生来的新药物还在给人们带来不断的惊喜。
紫杉醇的后继者
上面提到的法国科学家Potier,在探索用10-DAB合成紫杉醇的过程中,发现一个名叫RP5676的中间产物,距离紫杉醇还有两步。
Potier在对这个中间产物进行活性测试时,发现RP5676比紫杉醇有着更强的微管蛋白结合能力[13]。
这个中间产物,后来就成为了赛诺菲的抗癌药,多西他赛(Docetaxel)。多西他赛跟紫杉醇结构上非常相似,有着相同的母核,只是
有两个基团跟紫杉醇不一样[13]。
1996年,多西他赛获得美国FDA的上市批准用于治疗晚期乳腺癌,后来又被批准用于治疗非小细胞肺癌、前列腺癌、胃腺癌等。
尽管紫杉醇和多西他赛的疗效很好,但它们都有一个弱点就是水溶性都很差,所以需要用有机溶剂进行溶解。紫杉醇用的是聚氧乙烯蓖
麻油,而多西他赛用的是吐温-80和乙醇。这些溶剂很容易刺激机体释放组胺,导致过敏。
所以,在用药之前,患者通常需要皮质激素及抗组胺药的预处理,以减轻过敏反应[14]。这无疑增加了患者的负担。
于是,一种叫做白蛋白结合型紫杉醇(Abraxane)的新一代紫杉醇药物应运而生。
Abraxane利用人源白蛋白作为载体,可直接给药,无需用药前皮质激素和抗组胺药的预处理[14],可以说给患者带来了更好的体验。
2005年,Abraxane被美国FDA批准用于乳腺癌治疗。之后,又被批准用于非小细胞肺癌、胰腺癌的治疗。Abraxane也成了抗癌药市
场冉冉升起的新星。
回顾紫杉醇从诞生以来半个多世纪的历史,我们不得不赞叹大自然的神奇,可以给人类提供如此有效的抗癌药!也要感谢那些在药物研
发过程中不断坚持和付出的科学家们!
参考文献
[1] Hirsch J. An anniversary for cancer chemotherapy[J]. JAMA,2006, 296(12): 1518-20.
[2] Cragg GM. Paclitaxel (Taxol®): a success story with valuable lessons for natural product drug discovery and development[J].
Medicinal research reviews,1998, 18(5): 315-31.
[3] Wall ME, Camptothecin E. taxol[J]. Chronicles of Drug Discovery American Chemical Society, Washington DC,1993: 327-48.
[4] Wall ME, Wani MC. Camptothecin and taxol: discovery to clinic—thirteenth Bruce F. Cain Memorial Award Lecture[J]. Cancer
Research,1995, 55(4): 753-60.
[5] Wani MC, Taylor HL, Wall ME, et al. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and
antitumor agent from Taxus brevifolia[J]. J Am Chem Soc,1971, 93(9): 2325-7.
[6] Fuchs DA, Johnson RK. Cytologic evidence that taxol, an antineoplastic agent from Taxus brevifolia, acts as a mitotic spindle
poison[J]. Cancer treatment reports,1978, 62(8): 1219-22.
[7] Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol[J]. Nature,1979, 277(5698): 665.
[8] Rowinsky E, Donehower R, Rosenshein N, et al. Phase II study of taxol in advanced ovarian epithelial malignancies. Proc Am
Soc Clin Oncol; 1988; 1988. p. 136.
[9] Holton RA, Kim HB, Somoza C, et al. First total synthesis of taxol. 2. Completion of the C and D rings[J]. Journal of the American
Chemical Society,1994, 116(4): 1599-600.
[10] Nicolaou K, Yang Z, Liu J, et al. Total synthesis of taxol[J]. Nature,1994, 367(6464): 630.
[11] Li Y. Design, Synthesis and Evaluation of Novel Taxane-Based Anticancer Agents: State University of New York at Stony
Brook; 2011.
[12] Denis JN, Greene AE, Guenard D, et al. Highly efficient, practical approach to natural taxol[J]. Journal of the American Chemical
Society,1988, 110(17): 5917-9.
[13] Guenard D, Gueritte-Voegelein F, Potier P. Taxol and taxotere: discovery, chemistry, and structure-activity relationships[J].
Accounts of chemical research,1993, 26(4): 160-7.
[14] Miele E, Spinelli GP, Miele E, et al. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast
cancer[J]. International journal of nanomedicine,2009, 4: 99.
中国免疫接种发展史
在中国历史上,天花、麻疹、白喉、猩红热、鼠疫、霍乱、伤寒、痢疾等等病毒都曾肆虐一时,夺去了无数人的生命。为了战胜各种
病毒,世界各地的科学家们研制了各种疫苗,将人类从病毒的魔爪中拯救出来。
今天我们就来回顾一下,疫苗在中国经历了怎样的发展轨迹?
世界上通过免疫预防疾病的实践最早起源于中国。1727年俞茂鲲《痘科金镜赋集解》记载,明隆庆年间(1567年~1572年)安徽太
平县首创种痘法。
张璐《医通》(1695年)记载痘衣、痘浆、旱苗等法,并指出其“始自江右,达于燕齐、近者遍行南北”。此法后传至欧洲各国,导
致牛痘接种的发明。经过多年的努力,1979年世界卫生组织宣布已在全世界消灭了天花病。
在清朝,鼠疫在1860-1912年间猖獗,尤其是1910年发生在东北的鼠疫。
中国的“人痘”法,传入欧洲后变为“牛痘”法,并于1805年回传入中国。不过因药品价格过高,这类医疗措施只能做重点示范,离
全国普及尚远。
1919年3月,北洋政府成立中央防疫处,地址在天坛。中国以前从无防疫的专门机构,疫苗全靠进口,它是我国历史上第一个国家卫
生防疫和血清疫苗生产研究专门机构。1928年,中央防疫处隶属于南京国民政府。
中央防疫处成立后,宗旨系以防疫为主,研制和生产霍乱菌苗、牛痘苗、巴氏狂犬疫苗、结核菌素、白喉抗毒素、百日咳菌苗、鼠疫
菌苗、肺炎菌苗等。民国政府在一些省市设置卫生试验所,从事疫苗生产。民国时生产疫苗的私人厂家不多。
新中国成立后,在接收原民国中央防疫处和西北防疫处的基础上,中央政府扩建和新组建了卫生部六大生物制品研究所,分片包干当
时全国六大行政区(东北区、华北区、西北区、西南区、中南区和华东区)的流行性传染病的预防和控制,负责疫苗的研究和生产。
1959年,在昆明成立了中国医学科学院医学生物学研究所,主要研究生产脊髓灰质炎疫苗。建国初期还曾有大连生物制品研究所,后
来被撤销。
建国后的六大研究所包括:长春生物制品研究所、北京生物制品研究所、兰州生物制品研究所、成都生物制品研究所、武汉生物制品
研究所和上海生物制品研究所。
新中国成立后,全国普种牛痘苗。1962年,卫生部发布《种痘办法》,1963年发布《预防接种工作实施办法》,在广大城市对免疫
对象按免疫程序进行四种疫苗(卡介苗、脊灰糖丸、百白破、麻疹)的适时接种,在农村则主要开展冬春季的突击接种。
痨病是结核病的俗称,由于患者晚期身体重度消瘦,营养不良、贫血导致肤色苍白,所以结核病又被称为“白色瘟疫”,曾长期被视
为绝症。卡介苗是两位法国细菌学家卡默德和介兰在20世纪初发明的。中国卡介苗的接种始于1933年,由王良医生从法国引入,他在重
庆建立起中国第一个卡介苗实验室,开启了中国研究培育卡介苗的先河。
1986年6月20日,经国务院批准,由卫生部、国家教育委员会、全国妇联、广播电影电视部、经济贸易部、国家民委联合发布通知,
成立全国儿童计划免疫工作协调小组和每年4月25日开展全国儿童预防接种日活动,其近期目标是实现国家“七五”计划提出的1990年争
取“使全国免疫接种率达到85%以上”的目标。
二十世纪80和90年代,我国政府对小儿麻痹症的预防非常重视,1993年,国务院办公厅转发卫生部关于开展强化免疫活动消灭脊髓
灰质炎报告,党和国家领导人亲自参与强化免疫活动,给孩子们喂“糖丸”。“糖丸”全称为脊髓灰质炎减毒活疫苗。
国产乙肝疫苗是1975年研制出来的,研制者陶其敏是北大人民医院肝病研究所首任所长。根据国际惯例,疫苗首先要在大猩猩身上进
行检验。可是她一直没有找到合适的大猩猩,就在自己身上做试验。
第一代乙肝疫苗为血源疫苗,由于其生产能力等方面受到限制,逐渐被采用新技术也更安全有效的基因工程重组疫苗所替代。为了加
速控制乙肝,我国在从国外引进技术的基础上,于20世纪九十年代生产出了第一批重组乙肝疫苗。
在疫苗接种不断普及的同时,我国在疫苗研发领域也取得了喜人的成绩。
1994年2月,浙江医学科学院由毛江森院士领导的团队研制成功的国家重大攻关项目——“甲型肝类减毒活疫苗”,已逐步向大批量
生产发展,为控制甲肝做出了重大贡献。
流行性出血热是一种很凶险的疾病,且具有极强的传染性,死亡率可高达30-40%,我国是世界上“出血热”的最主要疫区。浙江省卫
生防疫站朱智勇教授,置个人凶险于不顾,历10多年科研创新之功,终于研制出举世瞩目的“出血热”疫苗,为人类有效地控制这种烈性传染病
的流行带来了希望。
2009年以来,甲型H1N1流感在全球广泛流行,为有效应对疫情,我国科学家在全球率先研制成功甲型H1N1流感疫苗,并使我国成
为第一个应用该疫苗的国家。
2007年,卫生部下发《扩大国家免疫规划实施方案》,将免疫规划疫苗扩展为14种,可预防15种传染病。在努力巩固计划免疫取得
的成果基础上,建立了较为完善的预防接种服务体系,预防接种服务能力显著提高。
接种疫苗是国际社会公认的预防和控制疾病最经济、最简便、最有效的措施。
今年是我国实施免疫规划政策40周年,在历代免疫规划工作者的共同努力下,我国和我省的免疫规划事业取得了辉煌的成就。
来 源 | 浙江健康教育
格列卫:一个神药的传奇——愿病者有其药
来源:我是科学家iScientist
徐峥与宁浩共同监制,徐峥亲自主演的一部喜剧电影《我不是药神》在全国院线上映。这部在点映阶段就已经口碑爆棚的电影可以
说是有笑点更有泪点,甚至有影评人称之为“年度迄今为止的最佳国产电影,没有之一”。
不过,电影终归是电影,而不是科普。电影要关注的是人物的抉择与命运,如果你想对电影中的“主角”格列宁有更多的了解,请听我
八一八这种来自神油国的神药背后的故事。
格列卫让血癌“慢”下来
可能有人会以为格列宁是种虚构的药物,但其实对抗癌药物稍有了解的人就会知道,片中的格列宁所“扮演”的必然是抗癌神药——格
列卫。它的欧洲商品名为Glivec,美国商品名为Gleevec,实际的药物分子名为伊马替尼(Imatinib)。
格列卫的确称得上是一种“神药”:病人对这种药物的响应率几乎为百分之百;长期服用不会产生耐药性;可以治疗的疾病范围不断
扩大。更重要的是,格列卫居然把生存希望不大的癌症变成了吃药就能控制的慢性病。当然了,还有它“神”一样的高昂售价。
为什么格列卫会这么神?
首先,得介绍一下格列卫主要治疗的这种癌症——慢性粒细胞性白血病(chronicmyelogenous leukemia,CML)。所谓白血病,即
是血癌。我们知道,癌细胞是积累了很多基因突变的细胞,能够无限增殖。具体到CML上,病人血液中出现基因突变的细胞是各种粒细胞。
粒细胞是白细胞中的主力,比如占比最高的中性粒细胞能够占到全部白细胞的40%到70%。中性粒细胞是我们免疫系统负责天然免疫的
主力军之一。当你的身体出现损伤时,中性粒细胞可以在一二十分钟之内到达损伤部位,吞噬入侵的病原体等异物。
血细胞主要都是在骨髓中“生产制造”出来的。在CML病人的骨髓中,造血细胞出现了异常,不受控制地生产出了大量的粒细胞。
如事情仅仅是这样,问题还不至于太过严重。但是当这种癌症发展到急变期之后,骨髓内的粒细胞急剧增多,挤压了正常造血细胞的生
存空间。
要知道,我们血液中的各种血细胞的寿命都是很有限的,比如红血球的寿命就是不到四个月的时间。因此,这些血细胞都需要时常补
充再生。那么当CML病人的正常造血细胞减少时,身体必然就会出现各种严重的症状,包括免疫机能的丧失,最终导致病人的死亡。
CML虽然带着一个“慢性”的名头,但绝不是可以掉以轻心的癌症。如果癌症是发生在具体的组织或器官上,手术切除或是有针对
性的放化疗通常就能解决问题。但CML却是发生在骨髓中以及血液中,根本无法用上述方式治疗。
在本文的主角“神药”格列卫现世之前,CML唯一有效的治疗方法就是风险较高的骨髓移植。这也是很多国产电视剧中常见的桥段,
其中的困难与危险恐怕大家都耳熟能详了。如果无法通过骨髓移植来治疗,那么CML的生存时间只有3到5年左右。能够采用的药物治疗
主要是服用干扰素,但也只能将其中20%到30%病人的生命延长1年而已。
格列卫出现之后,有不同的研究机构对服用格列卫的CML病人进行了多年的追踪调查,结果只能用“神奇”来形容。病人用药之后
的五年生存率高达90%左右,且死亡者的死亡原因大多与其所患癌症无关,只有1%左右的病人是死于白血病的恶化。可以说,格列卫
把一种恶性癌症变成了一种只需服药就可以控制的慢性病,总算是让CML中的“慢性”二字名符其实了。
费城染色体的交换术
为什么格列卫会有如此强大的药效呢?这就得从CML根源上的致病原因说起了。
1950年代末期,位于美国费城的宾西法尼亚大学医学院的病理学家PeterNowell在研究白血病病人的白细胞时意外发现,细胞核中
的染色体形态出现了异常。虽然我们总说性染色体分为X形和Y形,但其实我们另外那22对染色体全都长得像是字母X一样,只不过X的
两划之间打开的角度并不大,更像一个没写好的H。而Nowell却发现,有些病人白细胞中的染色体打开了一个大大的角度。
为了弄清楚这是怎么回事,Nowell找到了同在费城的福克斯·蔡斯癌症中心的一位研究染色体的博士生DavidHungerford帮忙。后者
进行了大量的观察和研究之后得出结论,这些白血病人的白细胞中出现异常的染色体是第22号染色体,不但X形分开的角度变得异常大,
而且染色体本身还变短了。于是,这种异常染色体就用两位发现者所在的城市进行了命名,称为“费城染色体”。
由于费城染色体的发现,白血病成为了人类历史上第一种找到了遗传异常证据的癌症。但是,费城染色体的成因此时仍然未知。又
经过了不同实验室几十年的研究,科学家们才对此有了较为深入的认识。实际上,22号染色体不是唯一出现异常的染色体,还有一个第
9号染色体也发生了异常,只不过是异常变长了。原来,第9号染色体长臂上的一部分,与第22号染色体长臂上的一部分进行了交换。结
果才导致前者变长,后者变短。
糟糕的是,两条染色体上发生交换的断点部位恰巧都在某个基因的内部,在第22号染色体上是BCR基因,而在第9号染色体上是
ABL1基因。于是,交换之后,在嵌合的22号染色体上出现了一个融合基因,也就是由两个基因连在一起形成的基因,其中保留了BCR
和ABL1的大部分,因此称为BCR-Abl基因。
我们知道,基因都是用来编码蛋白质的,这个BCR-Abl基因就会生产出来一种不存在于正常人细胞中的嵌合蛋白,称为BCR-Abl。
由于第9号与第22号染色体发生易位的具体位置并不是固定的,所以不同白血病人白细胞中所带有的BCR-Abl蛋白也不相同。其中分子量
为210 kDa(千道尔顿)的那种BCR-Abl蛋白就是引发90%的CML的罪魁祸首。
常开不关的分子开关
为什么一个BCR-Abl嵌合蛋白就能兴风作浪呢?因为这个嵌合蛋白恰巧是一个开关,一个控制着细胞分裂增殖的开关。
完整的Abl蛋白是一种酪氨酸激酶。所谓激酶,是一种能够在底物蛋白上添加一个磷酸基团的酶,这种过程被称为磷酸化。磷酸基团
是个很妙的家伙,它有着强烈的负电性,能够吸引一切带正电的东西。
这就好比在你的脚上本来有个磁正极,现在让你用手抓住一块强力磁铁的磁负极,你的手肯定就会一下子被拽到脚边上去了。蛋白质
发生磷酸化之后,往往也会发生带正电的部位与磷酸集团相互吸引的情况,于是就会发生显著的形状变化,进而影响到蛋白质的功能。
实际上,在我们的细胞里有很多由激酶组成的链路。激酶甲发生磷酸化之后被激活,就有能力去磷酸化激酶乙了。激酶乙发生磷酸化
之后被激活,就有能力去磷酸化激酶丙了。如此辗转,细胞里的信号就能被一直传递下去了。这样的信号通路有一个很形象的名字,称为
激酶瀑布(kinasecascade),或者也可以较为无趣地译为激酶级联反应。
当然,也有些激酶不是通过其他激酶来激活的,比如Abl就是这样一个激酶。它的头部有一个形如盖子的区域,结合在自己的酶活中
心上面,使得整体处于一种失活状态。Abl需要细胞中的其他蛋白质来打开自己的盖子,才能被激活。这是细胞“开关”Abl活性的调控
方式。
然而CML病人细胞内出现了BCR-Abl这种异常的融合蛋白。它的基因融合时,Abl靠后,恰好把具有抑制功能的头部盖子对应的半个
基因丢在了第9号染色体上,反而替换上了22号染色体上无用的半个BCR基因。于是,BCR-Abl中的Abl成为了一种永远处于开启状态的
激酶。相应的,带有BCR-Abl的细胞也就处于一种异常的不停分裂增殖的状态,成为了癌细胞。
格列卫的诞生
搞清楚了CML的致病原因,咱们的主角格列卫终于快要登场了。
在上个世纪的后半叶,癌症几乎就等于是不治之症。有一小部分幸运的病人可以通过手术和放化疗得到基本治愈,但大部分癌症患者
就没这么幸运了。对于后者,医生所能做的无非两点:一是为病人减轻痛苦,二是有限地延长病人的生命。不过,当时有一些医生已经不
再满足于此了。
随着生物学对于细胞的认识逐渐微观到了基因和蛋白质的层次,科学家们已经知道了一部分癌症的确切起因,比如CML,因而可以去
寻找更有针对性的治疗方法了,而不再是用普通的化学药物去无差别地杀伤所有细胞。
具有医学和生物学双学位的BrianDruker教授就是这样一位癌症研究者。1988年的时候,他刚刚来到俄勒冈健康与科学大学建立自己
的实验室,一位瑞士制药公司Ciba-Geigy的科学家Nicholas Lydon博士找到了他,希望与他合作开发一种有针对性的抗癌化学药物。于
是Druker告诉Lydon,最佳的选择就是针对CML这种癌症进行研究,因为它的分子机制在当时已经基本搞清楚了。更重要的是,导致这
种癌症的BCR-Abl蛋白是基因异常融合的结果,所以不会出现在正常细胞中。这样一来,针对BCR-Abl开发的药物就不会对正常细胞起
作用。
就这样,Druker和Lydon展开了以CML病人细胞中的BCR-Abl蛋白为靶标的合作。很快,Lydon搞到了一个以炎症为治疗目标而构建
的小分子库。他们用这个库中的小分子一一进行测试筛选,最终找到了一个编号为STI571的化合物,能够有效杀灭CML病人异常的粒细胞。
此时找到的化合物还不等于药物,而是通常被称为先导化合物。因为这个化合物虽然对目标细胞有杀灭作用,但是难免在进入人体之
后发生各种各样的情况,比如被人体迅速代谢掉,或是被人体的解毒机制处理成没有药物活性的小分子,或是对人体产生剧烈的毒性。所
以,先导化合物还需要进行进一步的改造,并经过动物实验和临床实验的验证。遗憾的是,很多化合物无法通过这些实验,最终也就不
能成为药物。STI571很幸运,经过简单改造之后就进入了动物实验阶段。
在当时的1990年代,人们对于癌症药物的研究是比较绝望的。美国有一位癌症研究专家就曾经开玩笑说:“人类对于癌症的研究史就
是一部小鼠癌症的治愈史。”言下之意是说,很多药物在动物实验时的效果都很好,能够有效治愈动物身上的癌症,但是对人类癌症却完
全无效。
格列卫的情况却恰恰相反。它在很多种类的动物身上都表现出了一些不良的副作用,特别是对于狗来说,严重的肝脏反应几乎杀死了
参与试验的狗。眼看这个项目就要胎死腹中的时候,在猴子身上的试验却取得了成功,才得以让格列卫的故事延续。
然而,好事多磨。就在格列卫即将进入人体临床实验之际,公司层面发生了巨大的变动。Lydon所在的Ciba-Geigy公司与另一家制药
公司合并,组成了如今国际制药界的巨头,瑞士诺华公司(Novartis,电影中的“诺瓦公司”)。Lydon也在这场动荡中离开了公司,而
合并后的新公司似乎对这个还没有上临床的项目不太感兴趣。
好在,Druker是一个非常有毅力的人,锲而不舍地说服了诺华的CEO,得以让格列卫进入临床阶段。Druker也的确得到了幸运女神
的垂青,他们的药物在一期临床试验中表现出了极好的药效,所有参与试验的CML病人对于格列卫百分之百产生了响应。如此高的响应
率是此前的抗癌药物研发中未曾有过的奇景。
很快,这种尚在试验之中的“神药”格列卫的名声在CML病人间流传开来。要求加入这项临床试验的请求如同雪片般堆积到了诺华
CEO的面前。于是格列卫的第二期、第三期临床试验得以顺利展开。
最终,2001年5月,美国食品药品管理局(FDA)批准格列卫作为一种治疗CML的药物上市销售。此时距离Druker与Lydon首次会
面过去了13年,距离诺华向FDA提交药品上市申请过去了两年半。
理性的胜利
格列卫的上市销售在社会上引发了巨大的反响。就在它获批的同一月,便登上了美国《时代周刊》的封面,称其为革命性的药物,并
被喻为抗癌战争中的新武器。
要知道,格列卫所专门治疗的CML并不是一种高发性的癌症。根据非官方机构2014年的统计,在我国大陆地区,CML的发病率在成年
人中不及十万分之一。西方国家的发病率略高一些,但也不超过十万分之二。
即便格列卫几乎把这种癌症的死亡率降到了零,但也远远谈不上攻克癌症。那么为什么人们会对它如此重视呢?
首先,作为一种主要治疗CML的抗癌药物,格列卫实在不负“神药”之名。前文已经说过,格列卫能够使CML病人的五年生存率从
30%左右,提高到接近正常人群水平的90%,把癌症变成慢性病。
其次,格列卫不像其他化疗药物那样会产生耐药性。也就是说,癌细胞无论如何突变,也无法躲避格列卫的杀伤作用。
另外,CML病人对格列卫的响应率很高,接近百分之百。要知道,相当多的抗癌药物只能对相应癌症病人中的一部分有效,对同样患
此病的其他病人则完全没有效果。更为神奇的是,这种靶向BCR-Abl的药物也显现出了“脱靶”的情况,对除CML之外的多种癌症都表现
出了良好的疗效。
目前,格列卫已经被FDA批准用于十种不同癌症的治疗。甚至还有研究者报道,格列卫对于糖尿病也会产生一定的疗效。
从生命科学研究的角度来讲,格列卫是人类历史上第一种理性设计的药物。在格列卫之前,药物研发套用一句咱们的俗话就是“打哪
儿指哪儿”。比如上世纪六七十年代的抗生素大发现时期,基本上就是去那些偏门的地方寻找偏门的微生物,以期能够在它们的细胞中找
到一种全新的抗生素。这个过程的本质是“非理性的”。甚至还有很多药物最终上市治疗的疾病根本不是药物公司最初研发它时希望去治
疗的疾病,比如蓝色的小药丸“伟哥”就是一个典型的例子。
而格列卫则不同,堪称“指哪儿打哪儿”的典范。它的研发从一开始就指向了CML,并且从疾病的基因变异到突变蛋白质再到抑制小
分子,一步一步都有理性的依据,因此称为理性设计的药物。Druker和Lydon也因为他们在理性药物设计方面开创性的工作而在2009年
获得了被誉为“生物学和医学诺奖风向标”的美国“拉斯克奖”。
实际上,今天的药物研发甚至可以做得更好。
就在格列卫上市销售之后的第二年,才由研究蛋白质三维空间结构的结构生物学家报道了格列卫与它所抑制的BCR-Abl蛋白紧密结合
在一起的复合物结构,切切实实“看”到了格列卫上的每个原子是如何与BCR-Abl结合的,是如何发挥功能的。至此,格列卫的科学故事
才算是彻底完整了。
然而,如果我们把思路倒过来:要是我们能够先得到BCR-Abl的结构,是否可以根据它的活性中心的形状和带电情况来设计一种药物
小分子,填进去,堵死它,来抑制它的功能呢?如果能做到的话,就把格列卫研发过程中最后一个“瞎猫碰死耗子”的环节——也就是对
小分子库的筛选——也变成“理性设计”了。这种方法就叫做“基于结构的药物设计与改造”。
新世纪以来,的确有很多研究机构和药物公司都在用这条思路来设计研发药物分子。只可惜,成功的案例凤毛麟角。应该说,人类对
于微观世界里蛋白质分子与其他小分子相结合时所发生的事情,仍旧了解得十分有限,这就给相应的药物设计带来了很大的阻碍。但是
毫无疑问,基于结构的药物设计是未来药物研发的一个重要方向。
转载自:新浪科技> 科学探索> 生命医学 > 正文
历史:发现胰岛素,胰岛素的发现发展历史
在亚洲,四十岁以上的人群,十人中就有一人患有糖尿病。著名医学期刊《新英格兰医学杂志》公布的最新的调查结果显示,糖尿病
在中国的发病率达到9.7%。现今,在全世界有超过3亿糖尿病患者。
胰岛素成功地用于人体试验
实验初步成功了,但仍然有一大堆困难摆在面前:首先是胰岛素不够用。用先前的方法提取胰岛素,程序复杂耗时。其次,提取液必
须近一步提纯,才能应用于临床病人。此时的麦克劳德改变了态度,他不仅本人直接参与实验,还动员了他的助手生化学家科立普加入。
不久,科立普发现酸化酒精能够抑制胰蛋白酶的活性,可以从动物的胰腺中直接获取提取物,并保存下来。用这种方法可以获取纯度
更高的胰岛素。
1922年1月,胰岛素第一次成功地用于人体试验。它被注射到一个14岁的糖尿病患者的体内。这个14岁的男孩叫兰纳德·汤姆森。当时
他正在接受饥饿疗法,体重已经剩下不到30公斤,估计几个星期后就会死去。
1922年1月11日,多伦多大学的医生给兰纳德注入了班廷团队提取的胰岛素。半个小时后,男孩的血糖值就下降了25%。12天以后,
医生开始给他连续注射,兰纳德的血糖指标下降了75%,尿糖近乎完全消失,精神、体力明显恢复。之后,医生们又在几个成年糖尿病人
身上进行了临床实验,也获得了惊人的效果。胰岛素对糖尿病的疗效,已经确凿无疑了。
胰岛素的提取,给成千上万单纯性缺乏胰岛素的病人带来了福音。只要按时按量注射胰岛素,尽管不能治愈糖尿病,但可以大大缓解
糖尿病的症状,不仅能延长患者生命,而且能极大地改善生活质量。
大量制造胰岛素
当时获得胰岛素的方法就是从屠宰场拿到冷冻的牛和猪的胰脏,再从磨成粉的胰脏中提纯胰岛素。班廷的团队只能满足很少一部分病
人的需求。现在,如何大量制造胰岛素是摆在班廷面前的一个难题。
当班廷发现胰岛素的消息轰动世界的时候,一对丹麦夫妇正在美国度假。克罗格因是哥本哈根大学的一位教授,曾获得诺贝尔生理学
或医学奖。他的妻子玛丽是一名医生,也是一名糖尿病患者。凭着职业的敏感,他们意识到这个将会改变成千上万糖尿病患者命运的发现
也将改变他们的生活。
此前,美国的礼来公司已经获准大规模工业化生产胰岛素。但欧洲市场和全球市场的需求远非一个公司可以满足。经过谈判,最终,
他们获得了在丹麦生产胰岛素的许可权。回到丹麦后,克罗格因夫妇创办了一家公司,专门从事胰岛素的大规模工业生产,这就是现今全
球最大的胰岛素制造商诺和诺德公司的前身。1923年诺和诺德制造的胰岛素开始在市场上大量销售,全世界数百万的患者获得了生存的机
会,这其中也有克罗格因的妻子玛丽。
糖尿病是一种古老的疾病
公元前1550年,古埃及人书写在纸莎草上的文献上面,记载着一种多饮多尿的疾病。这是考古学上可追溯的最早关于糖尿病的文字记
录。大约3000多年的时间里,世界各地的人们都在试图用他们各自的语言描述糖尿病。
古代中医把糖尿病称为“消渴病”,因为病人有多食多饮多尿和体重消减的症状,仿佛是饥渴所致。公元前400年,中国的《黄帝内
经》中就有“消渴”病的记载。
糖尿病人的尿液里可以检测出葡萄糖,这意味着病人血液中的葡萄糖没有被有效地分解,以提供身体所需要的养分和能量。由于血液
中的葡萄糖不能有效地分解,一方面肌体无法得到充足的养料和能量,体重下降,病人身体日益虚弱。另一方面体内的脏器和组织受到高
血糖的腐蚀和侵害,逐渐丧失功能。糖代谢的紊乱又进一步引起蛋白质、脂肪、水和电解质等一系列代谢紊乱综合征。因为免疫功能减弱,
糖尿病人容易患感冒、肺炎、肺结核等各种感染疾病,而且不易治愈。一旦患上“糖尿病”,可能发生的并发症遍及全身。在这些并发症
中,出现率最高的是视网膜病变、肾病和神经障碍,这就是“糖尿病”的“三大特征并发症”。
治疗没有突破性进展
糖尿病人血液中的葡萄糖为什么不能正常代谢,继而引发遍及全身的并发症呢?1889年德国科学家明可夫斯基与梅林进行动物实验,
他们切除了狗的胰腺,以观察胰腺是否是生存所必需的,却意外地发现这些狗出现了糖尿病。由此推论,糖尿病的病因与胰腺有关。
随着科学的进步,科学家们不断推进对糖尿病的研究,而每一个小小的进步都为后来者的研究奠定了基础。尽管如此,直到20世纪初,
糖尿病的治疗还是没有突破性的进展。在1914年关于糖尿病的文献上有这样的描述:“医院里满是糖尿病人,许多是儿童,他们会慢慢死
去。当时的疗法就是将他们送进医院,注射一些生理盐水。”直到1921年,医生们还在用饥饿疗法来延长糖尿病人的生命。
一旦患上糖尿病,这个病人的生存期也就是一两年。病人的血糖很高,严重的口渴,然后就是酮症酸中毒、昏迷,严重的营养不良,
消瘦,很快病人就会出现脱水,直至死亡。
实验初步成功了
真正揭开血糖代谢之谜,寻找到有效治疗糖尿病方法的是加拿大的外科医生弗雷德里克·班廷。班廷克服了技术难关,首次成功地提取
了胰岛素,被称为“胰岛素之父”。他和另一名科学家麦克劳德在1923年获得了诺贝尔生理学或医学奖。
1920年10月,加拿大安大略省的医学院里,外科医生班廷正在准备给学生讲授一堂关于糖代谢的课。他知道这是一个与胰腺有关的问
题,他翻了好几本教科书,对胰腺这个器官的生理讲得太少了。有许多问题,如果学生问起来,他无法回答。胰腺为什么跟糖尿病的发生
有关?在教科书上找不到答案。
班廷开始在各种医学刊物中寻求答案,他很快欣喜地发现了一篇相关的论文。这篇论文让班廷确信胰脏中一定含有能控制血糖的物质,
而这种物质极有可能会存在于胰岛之中。他立即产生了一个想法:从几只实验用狗的胰脏里分离出细胞,产生胰岛素进行纯化,然后用这
些纯化的胰岛素来调节病人的血糖。
为了实现这个大胆的构想,班廷来到了加拿大多伦多大学,找到著名的生理学家麦克劳德,希望得到他的指导和帮助。麦克劳德答应替班
廷找两个大四的学生来帮忙。这两个学生一个叫贝斯特,另一个叫瑙波尔,因为经费等问题,两个学生只能轮流工作。
1921年5月,班廷和贝斯特在多伦多大学的麦克劳德实验室开始了实验。两个月过去了,进展甚微。实施手术的19只狗中,因感染死
了14只。这样的感染在抗生素发现之前,几乎无法避免。班廷没有薪水,一切实验费用都需要自己支付。实验室条件很差,闷热潮湿。班
廷昼夜工作,废寝忘食,在物质和精神的双重压力下,几乎喘不过气来。
到了8月,实验终于初见成效。他们在10只糖尿病狗身上,共注射了75次提取液,收到了降低血糖和尿糖的效果。其中有两只狗,一
只活了20天,另一只活到了70天。在维持注射时,糖尿病狗能像正常狗一样生活;当停止注射时,糖尿病症状就很快出现。如此反复试验
多次,提取液的治疗效果显而易见了,这使班廷和贝斯特兴奋不已。
马克创造了一个奇迹
胰岛素的发现使人们得以对糖尿病病理进一步地深入研究。胰岛素是人体内惟一的降血糖激素。正常人体的胰腺可以分泌足够的胰岛
素。但有些人的胰腺有先天的缺陷,它完全不能分泌或者不能分泌足够的胰岛素使身体完成糖的代谢。这类型的糖尿病被称为1型糖尿病,
虽然可以发病于任何年龄,但患病者多是青少年甚至是婴、幼儿。
丹麦的马克在2岁时发现患有1型糖尿病。他的患病来自于家族的基因缺陷,他的几个哥哥姐姐都没能幸免。马克今年70岁了,68年
来,糖尿病一直伴随着他,这让人觉得不可思议。马克是幸运的,当时全球研究糖尿病和制造胰岛素最著名的三大机构有两个在丹麦。当
他被确诊为糖尿病时,胰岛素已经可以在丹麦的药房里买到了。马克的父母教会他为自己注射胰岛素。马克认为自己2岁的时候发现了糖尿
病,并不是坏事。他从小养成了注射胰岛素的习惯,这成为了他生活的一部分。
现在,马克身体相当健康,除了每天不间断地监测血糖和注射胰岛素,他还保持运动。先天的糖尿病似乎并没有太多地影响他的生活。
对一个1型糖尿病人来说,马克创造了一个奇迹。因为他注射胰岛素的时间已经长达68年了,而且这一时间还在不断延续。
为改进胰岛素而努力
自从班廷从狗的胰腺中成功地提取出胰岛素并用于治疗糖尿病,全球范围内,很多的医疗研究机构都在为改进胰岛素而努力。
1978年,美国的科学家们找到了利用基因重组生产出了和人胰岛素的序列完全相同的胰岛素产品,简称人胰岛素。几年后,欧洲的科
学家也制造出同样的产品,因为与人的胰岛素相同,减少了患者的不良反应,人们再也不用依靠动物胰脏,而是能生产出纯净的人胰岛素
了。
DNA技术带来了一系列新的突破。长效、速效的剂型相继出现了。家用血糖测量仪也有了改进,针头变得更细。还有新型的一次性针
筒,胰岛素笔、胰岛素泵等。
胰岛素泵形状、大小如同BP机。它模拟人体健康胰腺分泌胰岛素的生理模式,通过一条与人体相连的软管向体内持续输注胰岛素,俗
称“人工胰腺”。这种装置能够按照人体需要的剂量将胰岛素持续地推注到患者的皮下,保持全天血糖稳定,以达到控制糖尿病的目的。
本文转载自:糖尿病网 作者:tnbz.com
阿司匹林是如何被发现的
阿司匹林在医学上作用如此之大 那么它又是怎么被发现和运用的呢?
四千年前 ,古苏美尔人有了一项惊奇的发现。如果他们刮下某种特定树木的树皮,并吃下树皮,他们的痛就会消失。他们并不知道他
们所发现的这件事,注定要影响医学的未来方向。
苏美尔人发现的是现在被称为阿司匹林的这种药物的前身,人们发现柳树及野生植物中普遍存在阿司匹林的有效成分。这种成分也因
此注入到了各文化的医疗传统中,如苏美尔,古埃及,古希腊以及其他的文化等。
大约在公元前四百年, 希波克拉底 ,现代医学之父。第一个建议说咀嚼柳树皮能止痛,并用柳树叶制成的茶来缓解分娩之痛。但我
们花了两千多年才完全研究出其潜力,十八世纪中叶,一个名叫爱德华·斯通的英国人花了五年时间做实验,证明柳树皮压碎而成的粉末吃
下去可以治发烧。
又过了大概70年 ,一个叫约翰·毕希纳的德国药剂师,最终确认并提炼出使这一切成为可能的物质。一种叫水杨苷的化合物,之后,
医生们开始习惯性地运用柳树皮和其他富含水杨苷的植物。 如草菊来缓解疼痛,发烧 ,炎症。但是 ,对这一特定的化合物的确定意外地
开启了操纵其构造的可能性。
1853年, 一位法国化学家成功用化学方式人工合成这一化合物。发明了一种叫乙酰水杨酸的物质,之后在1897年 ,拜耳制药公司发
现了一种新方法。把乙酰水杨酸当作一种止疼用的化合物来销售 ,称其为阿司匹林。
这被广泛认为是首批合成西药之一,起初, 阿司匹林仅仅是拜耳公司的品牌名称。Aspirin(阿司匹林)的首字母A代表乙酰基,spir
指代草菊,草菊的植物学名称是榆绣线菊。
不久之后 ,阿司匹林变成乙酰水杨酸的同义词。当其影响力提升 ,人们发现阿司匹林不仅能缓解疼痛,还能治疗许多跟炎症相关的
毛病。如类风湿性关节炎,心包炎 ,它会感染包裹在心脏周围充满液体的膜以及川崎病。这种病会让血管发炎。
然而 ,尽管阿司匹林有药用价值,在当时科学家仍不知道它是怎么起作用的。20世纪60年代和70年代, 瑞典和英国科学家终于有所
发现。他们证明 ,阿司匹林阻止了某些被称为前列腺素的化学物质的生产。这种物质抑制了痛觉和炎症的传递。1982年 ,这一发现让研
究者们获得了诺贝尔医学奖。
随着时间的流逝 ,研究也发现了阿司匹林的风险。过度服用会导致肠内和脑内出血,也会引起瑞氏综合征。这是一种罕见且易致命的
疾病,会通过传染作用于小孩的大脑和肝脏。
20世纪末期,新的止痛药使阿司匹林的成功黯然失色。因新药有更小的副作用,例如醋氨酚和布洛芬。但在80年代 ,对阿司匹林好
处的更多发现激活了人们对它的兴趣。事实上, 1982年诺贝尔奖获得者也证明了阿司匹林能降低血栓素的生产速率。这种化学物质会导
致血小板的凝结,之后会导致血块的形成。一项有重大意义的临床试验表明对于服用阿司匹林的人来说,他们心脏病发作的几率降低了
44%。
如今 ,医生给心脏病或中风患者开出阿司匹林药物。因为它会降低血块在动脉里形成的可能性,而这些血块会供给心脏和大脑。更有
趣的是,有一种新兴的研究称阿司匹林会降低患癌和因癌致死的风险,特别是肠癌。
这也许是由于阿司匹林有抗血小板的作用,通过降低血小板的活性, 阿司匹林也许会降低某种帮助癌细胞扩散的蛋白含量。这些研究
将仅仅是一种止痛药的阿司匹林,转变成了一种潜在的救生治疗手段。
今天 ,我们每年大约要消耗1000亿阿司匹林药片,并且研究者们继续研究其新的作用。现今 ,多用途的阿司匹林已改变了现代医
学。考虑到其卑微的出身 ,仅仅是刮柳树皮而已,这实在令人震惊。
本文转载自 TED教育 ・ TEDEd
“超级传播者“——伤寒玛丽
什么是“超级传播者”呢?
世界卫生组织提出,把病毒传染给十人以上的病人被称为“超级传播者”。例如2003年“非典”时期感染一百多人的广州周姓
患者;造成北京大学人民医院和东直门医院大面积感染的的北京李姓患者等。
而在人类历史中,也的确出现过令人闻风丧胆“超级传播者”,更可怕的是,此人同时还是一个“隐性传播者”。
1906年的夏天,银行家亨利·沃伦在纽约度假时,一家11人中突然有6人染上了伤寒,这在他们居住的牡蛎湾富人区是非常罕见
的。毕竟伤寒是流传于欧洲贫民窟的噩梦,与他们这里相距遥远。
伤寒杆菌的传播途径是粪口传播(新型冠状病毒也有存在粪口传播风险),病菌可通过粪便等排泄物污染水或食物传播,也可
通过日常生活接触,苍蝇或蟑螂等媒介传播。
十九世纪末的纽约是穷人的地狱,每天有大量的粪便、垃圾堆积在街道,而底层民众便是污染的受害者。威廉·麦克尼尔在《瘟
疫与人》中提到,“仅就那些死于伤寒的穷人来说,如果传染疫病的虱子还没有把他们送往地狱,别的疾病也会很快把他们带走。”
银行家无法理解养尊处优的家人怎会突然患上伤寒,便聘请了伤寒研究专家乔治·索伯前来调查。乔治博士仔细对比了牡蛎湾附
近发生的几次伤寒疫情,排查了水源、食物、外来人口后发现,每次伤寒爆发时,总能看到同一个名字:玛丽·梅伦。
玛丽是个来自爱尔兰的单身女人,做得一手好菜,但巧合的是,她为雇主服务几个月后总会换个地方继续工作,而她的雇主们
往往不久后染上伤寒。
乔治博士怀疑是玛丽传播了伤寒杆菌,决定对其大小便样本进行检测。然而玛丽从未有过伤寒的症状,对博士的提议恼羞成
怒,电影《伤寒玛丽》中,前来取样的博士被愤怒的玛丽用烤肉叉戳地落荒而跑。
不甘心的乔治博士随即上报卫生部门,玛丽被制服并带回了拘留所,经过检测,玛丽体内存在大量活跃的伤寒杆菌,而她在烹
饪前从不严格清洗双手,病菌就这样被送上了餐桌。玛丽被隔离了两年,但在此期间她从未发病。
两年后,《纽约美国人报》刊登了对玛丽的报道,人们同情玛丽的遭遇,还有人认为这是政府对爱尔兰移民的歧视。尽管乔治
博士坚决反对,卫生局主管还是决定释放玛丽,只要她不再从事厨师工作。
坚信自己无辜的玛丽却并没有听从劝诫,在解除隔离五年后重操旧业,并再次引发一家妇产医院的伤寒爆发。这一次,没有人
再同情她,她被带回传染病医院,从此再也没有离开。玛丽一生直接传染了52例伤寒,间接传染不计其数,而她自己始终没有发病,最
后死于中风。
对于玛丽为什么不发病,我们可以从伤寒杆菌的生活史中略窥一二。
伤寒杆菌从媒介进入人体消化道后,通常会有一段7~14天潜伏期,这时候人体无任何症状。而随着细菌不断增殖和侵入血液,释
放的细菌脂多糖会引起人体免疫细胞应答,剧烈的免疫反应引起持续高热,导致人体中毒,腹泻、肠道穿孔并伴随特殊的玫瑰疹的出现。
病人从潜伏期就能通过粪便排菌,病后2~4周排菌最多,有的甚至排菌超过3个月。
潜伏期的长短受到感染细菌数量以及个体免疫力的影响。玛丽这样的细菌携带者体质特异,伤寒杆菌在体内免疫细胞的围攻下失
去伤害宿主的能力,所以玛丽并没有什么特别的症状。但伤寒杆菌仍然可以在人体内低水平地增殖并随着人体排泄物排出体外,污染
周围环境。
玛丽的故事并没有随着她的死而消亡,直到今天,美国人有时还会以开玩笑的口吻称患上传染病的朋友为“伤寒玛丽”。尽管我
们可以推测,在那个卫生意识淡漠的年代,一定不只有一个这样的病菌携带者,但玛丽的传奇经历和她富有反抗性的斗争过程,使得
“伤寒玛丽”成为传染病史上臭名昭著的案例。
个人权利和公众健康的抉择依然在不断引发讨论。对于成为瘟疫传播者的人来说,只要不是像玛丽一样拒不承认或者故意去散播
疫情,他们同样是被命运捉弄的普通人,同样值得同情。
人类与瘟疫的斗争已经持续了几千年,历史告诉我们,疫情总会过去,希望好消息越来越多,希望我们很快都能放松地走在灿烂的
阳光下。
参考:
百度百科词条:伤寒玛丽
维基百科词条:Typhoid Mary
威廉.H.麦克尼尔(WilliamH.McNeill) 《瘟疫与人》中国环境科学出版社
李兰娟《传染病学》 北京人民卫生出版社
欣正人《瘟疫与文明》 山西人民出版社
王晓华 疾病命名中的错误和偏见 深圳特区报2015-10-20
本文转载中国科学院微生物研究所官网
幽门螺杆菌的发现史与防治
幽门螺杆菌是一种螺旋形、微厌氧、对生长条件要求十分苛刻的细菌,是目前所知能够在人胃中生存的唯一微生物种类。幽门螺杆
菌虽然在1983年才首次从慢性活动性胃炎患者的胃黏膜活检组织中分离成功,但其实在这之前就已经有许多的学者为此作出过很多研
究。
1892年,意大利病理科医师Giulio Bizzozero在对狗的胃腺和壁细胞处的显微镜观察,证实哺乳动物胃内存在种螺旋体样微生物定
居。也是最早认识到胃内存在有细菌学者。
1896年,Salomon也证实在狗和猴子的胃、小肠溃疡部位亦存在螺旋体样微生物。
1919年,微生物学家 Kasai和Kobayashi在发现了猫胃粘膜中的螺杆菌。
1906年 Krienitz和Luger通过人体解剖首次报道人胃内有螺旋体微生物定居。
1924年Luck和Seth发现人胃上皮细胞内存在相当高的尿素酶活性,从而间接证明人胃内存在细菌定。
1939年,Stone Freedberg和Doenges通过对242例尸体解剖发现大约43%的尸解胃中存在螺旋体样微生物,1940年他们观察到
了溃疡患者胃活检标本上的螺旋体(Spirochete), 但论文发表后却被医学界所否认。因为当时流行的观点认为:胃内是高酸环境,因
而是一个无菌器官,任何细菌都不可能在胃酸中存活。所以这项研究遭到怀疑、否定。并且还认为,那些组织学检查所发现的胃粘膜上
存在细菌的报道是由于检查时标本被污染的原故。
澳大利亚的Adrian Lee在1968年就在洛克菲勒大学成功培养出螺杆菌,然而他当时却没有意识到该细菌属于新的种类,与诺贝尔奖
擦肩而过。
在上个世纪70年代,我国的老一辈医学家就利用通过口服痢特灵及青霉素等药物治疗胃炎,就间接说明那个时候人们已经认识的胃炎
不单纯是胃酸的缘故,还与胃内细菌感染相关,只是缺乏进一步研究,错失幽门螺杆菌导致消化性溃疡这一重大医学发现。
直到1983年,澳大利亚临床医生Marshall和Warren才首次从慢性活动性胃炎患者的胃黏膜活检组织中成功分离出幽门螺杆菌,当时
他们为这种细菌命名helicobacter Pylori(hp)。为了提供更确切的证据来证实幽门螺杆菌感染是胃疾病的直接致病因素,Marshall于
1984年7月自己进行了一次吞服该菌的人体自愿者试验。Marshall的这一实验成功证实了幽门螺杆菌感染确实可引起急性胃炎。
Warren和Marshall不是第一个发现幽门螺杆菌的人,但他们依然走上科学的求真之路,甚至不惜以自身为实验对象,亲身证明了幽
门螺杆菌确实可能导致胃炎的发生。为表彰发现幽门螺杆菌以及该细菌与胃炎和消化性溃疡的关系,Marshall和Warren被授予2005年诺
贝尔生理或医学奖。
2017年10月27日,世界卫生组织国际癌症研究机构公布的致癌物清单初步整理参考,幽门螺杆菌(感染)在一类致癌物清单中。胃
癌是全球最常见的恶性肿瘤之一,预防和控制胃癌已日益引起人们的关注。研究指出,幽门螺杆菌生存于人体胃幽门部位,是最常见的细
菌病原体之一。据统计,世界有多半人口受到过幽门螺杆菌的感染,中国是幽门螺杆菌感染大国,一般人群中幽门螺杆菌的感染高达50%-80%。
医学家们认为,彻底消灭幽门螺杆菌并非难事,90%的细菌感染者经过1~2周治疗后,体内的幽门螺杆菌往往能被消灭殆尽。他们建
议,应当进行全民普查,至少应该对接受过胃部手术、有过胃病、或亲属中有过胃癌的人进行幽门螺杆菌的检查,并对感染者进行杀菌治
疗,这样有望控制胃癌。
幽门螺旋杆菌感染预防的关键是把好“病从口入”这一关。要做到饭前便后洗手,饮食尤其是进食生冷食品要讲究卫生,集体用餐时
采取分餐制是明智的选择,家里有幽门螺旋杆菌病患者时应该暂时采取分餐,直至完全治愈。幽门螺杆菌感染在人群中普遍存在,但只有
很少人患病。因此大家不必对其产生恐慌,对于带菌者也不需采取特殊的隔离措施。只要养成良好的饮食卫生习惯,可以减少幽门螺杆菌
感染的发生。
一次幸运的过失,弗莱明发现了救了数千万人性命的青霉素
相信小时候打青霉素针的那种恐惧,很多人都还记得。打青霉素需要打两针,一针是皮试针,第二针才是青霉素针。那时候医生把两
小瓶药一兑,慢悠悠的就过来了,虽然打针也不是特别痛苦的事情,但想到的时候,难免还是会心里发紧。
青霉素主要是消炎类的抗生素,可以说青霉素救了无数人的命,所以,这样的医学发明相比其它的发明而言,就显得更伟大。
发现青霉素的人叫亚历山大·弗莱明,而他发现青霉素,则是源于一次幸运的过失。
1914年,开打第一次世界大战的时候,弗莱明是皇家陆军医疗队的队长,当时他到了法国,发现很多士兵受了伤,伤口一受感染,
基本上就只能用消毒水消下毒。
但是在当时消毒水有个坏处,就是把坏的细菌杀掉了,同时把人体有用的细菌也杀了,所以不怎么太有用,那时候死掉的人非常多。
这种坏细菌就是常见的葡萄球菌,譬如谁受伤了,如果不及时处理,没过多久就会发烧,就是葡萄球菌在作怪。所以弗莱明就希望找
到一种东西,能把葡萄球菌杀掉,还不伤害对人体有用的细菌。
这一找就找了十多年,到了1928年。这一天,弗莱明出去度假去了,但是忘了自己还在实验室培养葡萄球菌,器皿的盖子还没盖,
这下细菌就全部暴露在空气中。
等三个星期后,弗莱明度假回来,发现本来要培养的葡萄球菌最后头上都长白毛了,就跟我们出差几个星期,东西没放进冰箱,结果
面包啊、饭啊都长毛了之类的。
弗莱明看到这情况,肯定怪自己粗心大意啊,本来要把这些没用的东西扔掉,结果发现这些葡萄球菌和空气接触,长出了青绿色的霉
菌后,最后葡萄球菌反而不见了,离这种青绿色霉菌越近,葡萄球菌越少。他就想,这霉菌身上一定有对葡萄球菌有害的物质。
所以,就因为这么一次过失,反而幸运的让弗莱明发现了青霉素,这就叫踏破铁鞋无觅处,得来全不费功夫。
就这么着,亚历山大·弗莱明成了世界上最伟大的发明家之一,获得了诺贝尔医学奖,获得140多项荣誉,他的这项发明伟大到被公认
为是救命药,让人类的普遍寿命延长了24岁。
关于青霉素还有这么一个故事:说青霉素发现后,英国的首相丘吉尔得了肺炎,那时候得肺炎基本上就很难扛得过去,没办法了,医
生们说,要不就用这个新发明的青霉素试试。
丘吉尔可是首相,怎么会同意拿自己当小白鼠呢,坚决不行!最后,别人告诉他,研究这项发明的人叫亚历山大·弗莱明,一听到这个
名字,丘吉尔马上就同意了。
难道他们之间还有什么渊源吗?是这样的:有这么一天,一个农夫在田地里干活,他发现有人喊救命,就赶紧跑过去,结果发现,不
是有人掉水里了,是有一个小孩掉在粪坑里了。救人要紧啊,他二话没说,一捏鼻子就跳下去了,死活把人救了上来。
这事算过去了,但几天后,有个大富豪坐着大马车就来了,送了一份厚礼,来感谢这个农夫。原来农夫救的小孩是这个大富豪的儿
子,也算是知恩图报。但是这个农夫无论如何都不收大富豪的礼,他认为这是应该的,换做谁都会救的。
大富豪也没了办法,他就说,那这样吧,你救了我儿子,我也没什么报答。我也帮下你儿子,你让你儿子跟我走,我让他受最好的教
育,相信他成才以后你也会感到骄傲的。这么一说,农夫觉得可以,毕竟也不想看到儿子和自已一样当农民,就同意了。
最后,农夫的儿子就上了圣玛利医学院。到这儿大家都听出来了吧,农夫的儿子就是亚历山大·弗莱明,富翁的儿子就是首相丘吉尔,
所以丘吉尔一听,是自已恩人的儿子发明的青霉素,就同意了。
就像现在的百科上,对于亚历山大·弗莱明,也只是青霉素的发现者。
1940年,弗洛里和钱恩才正式在小白鼠身上,看到青霉素的疗效。他们分别取了8只小白鼠,给每一只注射了致死量的链球菌,给另
外4只注射了青霉素,几个小时后,打了青霉素的活嘣乱跳,没打青霉素的死掉了,小白鼠试完了才用在人的身上。
在二战末期,青霉素横空出世,迅速扭转战局,也救了数百万人的性命。青霉素也和原子弹、雷达一起,被称为二战最伟大的三项发
明。
相信在今天,我们每一个人都得感谢青霉素的到来,因为他一定救过你或者你亲人的命。
敬佩!屠呦呦回顾青蒿素发现艰难历程
2015年10月5日,诺贝尔奖委员会把诺贝尔生理或医学奖授予屠呦呦等三名科学家。2011年屠呦呦获拉斯卡奖之后,在《Nature》
撰文回顾了青蒿素发现的艰难历程。让我们我们瞻仰一下这份来之不易的诺奖起源。
约瑟夫·戈尔斯坦在这本杂志里提到了,创造(发明)和揭示(发现)是生物医学进步的两条不同的路径。作为一名植物化学家,我很有幸
地在这两条道路上都有所收获,尤其是在20世纪60年代到80年代这段日子里。1955年我毕业于北京医学院药学系,此后被分
配到中医研
究院(现中国中医研究院),从事中药研究。1959年到1962年之间,我参加了一项中医的培训课程,此课程主要是针对有西医背景的专业
人士开设的。两年半的培训让我发现了中医的奇妙和可贵,以及站在人类和宇宙的高度的哲学思辨之美。
一、青蒿素的诞生
疟疾是由恶性疟原虫引起的疾病,几千年来一直威胁着人类的生命安全。在20世纪50年代国际消灭疟疾的尝试以失败告终之后,疟
疾再度肆虐。这很大程度上归因于寄生虫对当时的抗疟药物,如氯喹,产生了抗药性。1967年,在“523办公室”领导下,中 国启动了
抗疟项目。我所在的研究院很快参加到了这个项目之中,并委任我为疟疾研究小组组长。该小组由植物化学和药理活性研究员组成。我们
这群年轻人开始了研究如何从中药里提取和分离抗疟有效成分。
工作的第一阶段,我们研究了超过2000种的中药,发现了其中的640种可能有抗疟效果。我们用小鼠模型评估了从大约200种中药里
获得的380种提取物。然而,过程并没有那么顺利。想要有重大发现谈何容易。
一份青蒿提取物给研究工作带来了转机。青蒿提取物很好地抑制了寄生虫的生长。然而,这个发现并没有在之后的实验中重复出来,并且
与此前文献中记载的有冲突。
为了找到合理的解释,我们翻阅了大量的文献。唯一一篇关于使用青蒿减轻疟疾症状的文献出自于葛洪的《肘后备急方》。文中提
到:“青蒿一握,以水二升渍,绞取汁,尽服之。”(图1)这句话给了我灵感。我们传统的提取方法里的加热步骤可能会破
坏药物的活
性成分。在较低的温度中提取可能有助于保持抗疟活性。果然,在使用较低温提取方法之后,提取物的活性得到了大幅提升。
明代(公元1574年)葛洪著作《肘后备急方》右图第五列“青蒿一握,以水二升渍,绞取汁,尽服之”。
随后我们把提取物分离为酸性和中性的两部分。终于,在1971年10月,我们获得了中性无毒的提取物。这份提取物对伯氏疟原虫感
染小鼠和食蟹猴疟原虫感染的猴子有着100%的疗效。这个结果标志着青蒿素发现上的突破。
二、从分子到药物
在文化大革命期间,是没有办法做新药的临床试验的。为了疟疾病患的福祉,我和我的同事们勇敢地成为了青蒿素的第一批志愿受试
者。在确认了青蒿素提取物对人体是安全的情况下,我们前往海南省,在患有间日疟原虫和恶性疟原虫的病人身上试验该提 取物的有效
性。
临床试验的结果激动人心:经治疗的病人疟疾症状迅速消失,这些症状包括高烧和血液中的寄生虫数量。而采用氯喹治疗的病人却没
有这些临床收益。
20世纪50年代,在中医研究院中药研究所任研究实习员的屠呦呦(前右)与老师楼之岑副教授一起研究中药。
积极的临床结果指引着我们再接再厉。我们接着分离纯化了青蒿的抗疟有效成分(图2)。1972年,我们得到了提取物中的活性成分,分子
量为282道尔顿的无色晶体,分子式是C15H22O5,熔点在56–157°C。我们将其命名为青蒿素。
《补遗雷公炮制便览》中彩色手绘青蒿(明代,公元1591年)与青蒿和青蒿提取物按照戈尔斯坦的观点,青蒿素的发现是我们科研
征程的第一步——揭示。我们接下来要进入下一步——创造,把自然界的分子转变为药物。
我们发现,在菊科蒿属的植物中,只有黄花蒿新鲜叶子中在花蕾期含有丰富的青蒿素。我所在的团队用的是北京本地的蒿属植物,青
蒿素的含量相对少得多。为了药物生产,我们迫切需要青蒿素丰富的蒿属植物。中国政府发起的寻找疟疾治疗药物的523项
目组是一个全
国范围的科技协作组织,项目组很快发现一种四川产的黄花蒿符合制药要求。
我们临床试验的第一种药物剂型是片剂。病人服用之后产生了不适感。在随后的研究中我们发现,这是因为旧压缩机器上生产出来的
药片很难分解。我们转而使用新的剂型——纯青蒿素胶囊,临床效果令人满意。本来看似关闭的抗虐新药开发的大门,再次
向我们打
开。
三、从国内到国外
除了药物生产和剂型的问题之外,我们面临的另外一个挑战是,如何让世界知道我们的发现。1975年,在中国科学院生物物理所的
一个科研团队的帮助下,我们确定了青蒿素的立体结构,是一种倍半萜内酯。
有关这个结构的论文,我们在1977年首次发表在中国的科学期刊上。同年,这个新分子和这篇论文立刻被美国化学文摘社引用。但
是,当时中国大的政治环境限制了任何关于青蒿素的论文发表。幸运的是,1979年,国家科委授予我们国家发明奖,表彰我 们发现青蒿
素和抗疟的功绩。
(a) 青蒿素的分子结构。(b) 青蒿素的三维结构模型,碳原子是黑色小球,氢原子是蓝色小球,氧原子是红色小球。
1981年,第四届化学药物治疗疟疾科学会议在北京召开。会议由联合国开发计划署、 世界银行和世界卫生组织发起。在一个针对热
带疾病的研究和培训的特别项目中,一系列关于青蒿素以及其治疗疟疾效果的报告引起了强烈反响。
作为会议的第一个发言者,我做了一个名为“青蒿素的化学性质研究”的报告。这些在报告中提及的研究,随后在1982年发表。20
世纪80年代青蒿素及其衍生物在中国治疗数千疟疾病人的疗效引起了全世界的关注。
1981年参加在北京召开的第四届化学药物治疗疟疾科学会议的代表合影。本文英文版作者屠呦呦在第二排,左数第四位。前排居中
的是季钟朴教授,时任中国中医科学院院长,在本次会议中对青蒿素的疗效做了公开的评价。
四、超越青蒿素
考虑到化学稳定性,二氢青蒿素一开始并没有被有机化学家看作一种有用的治疗药物。在评估青蒿素的各类衍生化合物时,我们发现
二氢青蒿素更加稳定并且比青蒿素的疗效好10倍。更重要的是,用二氢青蒿素治疗后,病人的疟疾复发率更低。通过酯化过
程向青蒿素
分子加入一个羟基基团增加了开发更多的青蒿素衍生物的可能性。
我所在的研究小组后来将二氢青蒿素发展为一种新的药物。过去的十年中,我和我的同事们已经开展了用青蒿素和二氢青蒿素治疗其
他疾病的研究。
在《青蒿素及其衍生物》一书中,我们总结了青蒿素的发现历史和我们对于青蒿素分子及其衍生物的研究成果。2005年,世界卫生
组织宣布了青蒿素联合疗法。青蒿素联合疗法现在广泛被采用,拯救了很多生命,其中很多是非洲儿童。由于能够有效地遏制
疟原虫配子
体细胞的活性,这种治疗方法显著地减轻了疟疾的症状。